Mole Concept
Understand how chemists measure matter using the mole. Learn conversion between particles, moles and mass using Avogadro's constant.
Master the foundation of chemistry — mole concept, stoichiometry, empirical formula, concentration terms and chemical laws. This chapter builds the numerical and conceptual base for the entire Class 11 chemistry syllabus.
Chapter Overview
Understand how chemists measure matter using the mole. Learn conversion between particles, moles and mass using Avogadro's constant.
Quantitative relationships between reactants and products. Includes balancing equations, limiting reagent and yield calculations.
Determination of chemical formulas using percentage composition, vapour density and molar mass relationships.
Important solution concentration units including molarity, molality, mole fraction and mass percentage.
Fundamental chemical laws proposed by Lavoisier, Proust, Dalton, Gay-Lussac and Avogadro explaining how elements combine.
Quick Formula Sheet
Number of moles: \[ n = \frac{m}{M} \] where \(m\) = mass of substance \(M\) = molar mass
Particles relation: \[ N = n \times N_A \] where \(N_A = 6.022 \times 10^{23}\)
Percentage yield \[ \% \text{Yield} = \frac{\text{Actual Yield}} {\text{Theoretical Yield}} \times 100 \]
Limiting reagent method \[ \frac{n_1}{a_1} = \frac{n_2}{a_2} \] where \(a\) represents stoichiometric coefficients.
\[ n = \frac{\text{Molecular Mass}} {\text{Empirical Formula Mass}} \]
\[ \text{Molecular Formula} = (\text{Empirical Formula})_n \]
Molarity \[ M = \frac{\text{moles of solute}} {\text{volume of solution (L)}} \]
Molality \[ m = \frac{\text{moles of solute}} {\text{mass of solvent (kg)}} \]
Concept Importance
Key Takeaways
Avogadro's number connects atomic scale quantities with measurable laboratory quantities.
The limiting reactant determines how much product can be formed in a reaction.
Convert percentages to moles, divide by smallest value, and obtain the simplest whole number ratio.
Volume changes with temperature, so molarity changes. Molality remains constant.
They reflect the reliability and accuracy of experimental measurements.
\[ \% \text{Yield} = \frac{\text{Actual Yield}} {\text{Theoretical Yield}} \times 100 \]
Study Strategy
Build a strong chemistry foundation by understanding the mole concept, stoichiometry and solution chemistry. Use the formula sheet and strategy above to revise the chapter efficiently.
Many scientists tried to explain chemical bonding interms of electrons, but in 1916, Kössel and Lewis independently gave the first successful explanation. Their idea of valence was based on the stable, unreactive nature of noble gases.
Lewis pictured the atom in terms of a positively charged ‘Kernel’ (the nucleus plus the inner electrons) and the outer shell that could accommodate a maximum of eight electrons.
Lewis postulated that atoms achieve the stable octet when they are linked by chemical bonds.
Lewis introduced electron-dot symbols to represent valence electrons. In these structures
Kössel, in relation to chemical bonding, drew attention to the following facts:
For example
| \(\mathrm{Na}\) | \(\mathrm{\rightarrow}\) | \(\mathrm{Na^++e}\) |
| \(\mathrm{[Ne]3s^1}\) | \(\mathrm{[Ne]}\) | |
| \(\mathrm{Cl + e}\) | \(\mathrm{\rightarrow}\) | \(\mathrm{Cl^-}\) |
| \(\mathrm{[Ne] 3s^2 3p^5}\) | \(\mathrm{[Ne]3s^3sp^6}\) or [Ar] | |
| \(\mathrm{Na^+ + Cl^-}\) | \(\mathrm{\rightarrow}\) | \(\mathrm{NaCl\ or\ Na^+Cl^-}\) |
The bond formed, as a result of the electrostatic attraction between the positive and negative ions was termed as the electrovalent bond. the electrovalence is thus equal to the number of unit charge(s) on the ion.
Kössel and Lewis developed an important theory of chemical combination between atoms known as electronic theory of chemical bonding. According to this, atoms can combine either by transfer of valence electrons from one atom to another (gaining or losing) or by sharing of valence electrons in order to have an octet in their valence shells. This is known as octet rule
A covalent bond is formed when two atoms combine by sharing one or more pairs of electrons. Unlike ionic bonding, where electrons are transferred, covalent bonding involves mutual sharing of electron pair so that each attains a stable outer-shell configuration. This type of bonding is most common between non-metal atoms whose electronegativities are comparable and whose tendency to lose or gain electrons completely is low.
Consider two hydrogen atoms approaching each other. Each hydrogen atom has one electron and requires one more to complete its duplet. When they come close, their electrons are shared, forming a common electron pair. This shared pair holds the nuclei together and results in the formation of a hydrogen molecule.
This principle extends to more complex molecules as well.
Depending on the number of shared electron pairs, covalent bonds are classified as:
As the number of shared pairs increases, the bond becomes shorter and stronger. Thus, a triple bond is generally stronger and shorter than a double bond, which in turn is stronger than a single bond.
Covalent bonds are often represented using electron-dot (Lewis) structures, where dots indicate valence electrons and a shared pair is shown either by two dots or by a line. These structures help visualize:
Although Lewis structures do not show the actual shape of molecules, they provide a useful first picture of how atoms are linked.
| Molecule | Lewis Representation |
|---|---|
| \(\mathrm{H_2}\) |
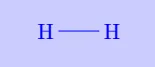
|
| \(\mathrm{O_2}\) |
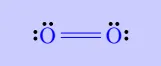
|
| \(\mathrm{O_3}\) |
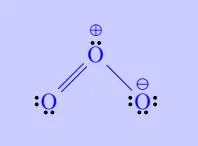
|
| \(\mathrm{CO_3^{2-}}\) |
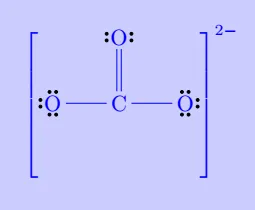
|
| \(\mathrm{NF_3}\) |
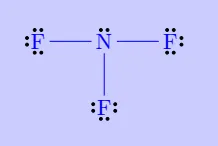
|
| \(\mathrm{HNO_3}\) |
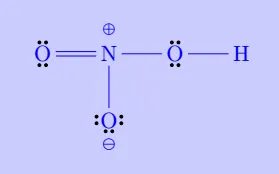
|
The formal charge of an atom in a polyatomic molecule or ion may be defined as the difference between the number of valence electrons of that atom in an isolated or free state and the number of electrons assigned to that atom in the Lewis structure. It is expressed as :
\[\mathrm{Formal\ Charge}=\mathrm{Valence\ electrons}-\mathrm{Nonbonding\ electrons}-\frac{1}{2}(\mathrm{Bonding\ electrons})\]
where:
Valence electrons are those of the free atom.
Nonbonding electrons are lone-pair electrons on that atom.
Bonding electrons are electrons shared in bonds.
This simple relation is applied atom by atom.
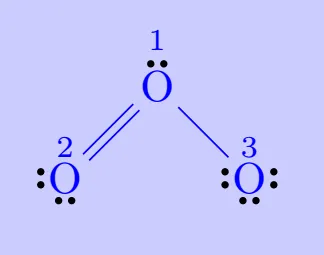
Let us consider the ozone molecule \(O_3\). The Lewis structure of \(O_3\) may be drawn as:
Formal Charge of central \(\mathrm{O}\) atom marked 1 \[ \begin{aligned} &=6-2-\dfrac{1}{2}\times 6\\ &=6-2-3\\ &=1 \end{aligned} \] Formal Charge of end \(\mathrm{O}\) atom marked 2 \[ \begin{aligned} &=6-4-\dfrac{1}{2}\times 4\\ &=6-4-2\\ &=0 \end{aligned} \] Formal Charge of end \(\mathrm{O}\) atom marked 3 \[ \begin{aligned} &=6-6-\dfrac{1}{2}\times 2\\ &=6-6-1\\ &=-1 \end{aligned} \]
Hence, we represent \(\mathrm{O_3}\) along with the formal charges as follows:
When multiple Lewis structures are possible, the preferred one generally satisfies these guidelines:
These principles help select the most realistic representation among several alternatives.
For example, in polyatomic ions and resonance systems, formal charge analysis clarifies why certain electron arrangements dominate even though multiple forms can be drawn.
Some molecules and ions cannot be represented adequately by a single Lewis structure. Instead, two or more contributing structures are written. Formal charge calculations show that these resonance forms often differ only in electron placement, not in atom positions.
The actual molecule is best described as a resonance hybrid, but formal charge remains essential for evaluating each contributing structure and understanding electron distribution.
Although both involve electron accounting, formal charge and oxidation number are conceptually different:
Formal charge is therefore more suitable for covalent molecules, while oxidation number is mainly used in redox chemistry.
Formal charge is a theoretical construct. It does not represent the true electronic charge on atoms and cannot predict molecular shape or bond strength by itself. However, it is extremely useful for:
The octet rule states that atoms tend to combine in such a way that each acquires eight electrons in its valence shell, thereby attaining a noble-gas-like configuration. This simple idea explains the bonding in many common molecules and ions. However, careful examination of several compounds reveals that the octet rule is not universal. While it serves as a useful guideline, it fails to describe bonding in a number of important cases.
These exceptions arise because atoms differ in size, available orbitals, and electronic requirements.
Some atoms form stable molecules even though their central atom has fewer than eight electrons in its valence shell.
A classic example involves boron compounds. In boron trifluoride (BF₃), boron is surrounded by only six electrons. Despite this apparent deficiency, BF₃ is a stable molecule. The reason lies in boron’s electron-deficient nature and its ability to accept electron pairs from other species, making such compounds chemically reactive but structurally viable.
Similarly, compounds like BeCl₂ also show incomplete octets around the central atom.
There are molecules that contain an odd number of total electrons, making it impossible for all atoms to achieve octets.
Nitric oxide (NO) is a typical example. Because the total number of electrons is odd, one electron remains unpaired. As a result, at least one atom must violate the octet rule. Such species are called free-radical molecules and are usually quite reactive due to the presence of the unpaired electron.
These molecules clearly demonstrate that octet completion is not always achievable.
Elements in and beyond the third period of the periodic table have, apart from 3s and 3p orbitals, 3d orbitals also available for bonding. In a number of compounds of these elements there are more than eight valence electrons around the central atom. This is termed as the expanded octet. Obviously the octet rule does not apply in such cases.
Examples include:
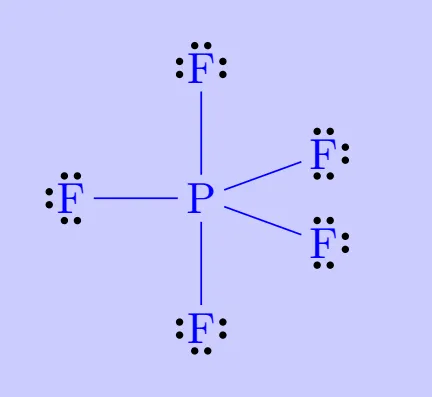
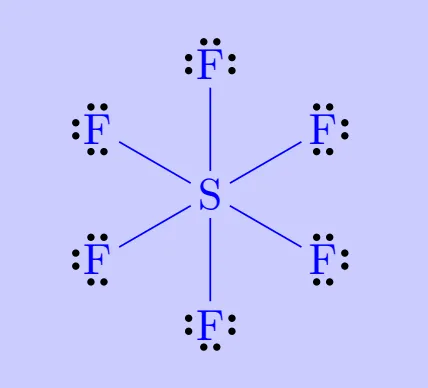
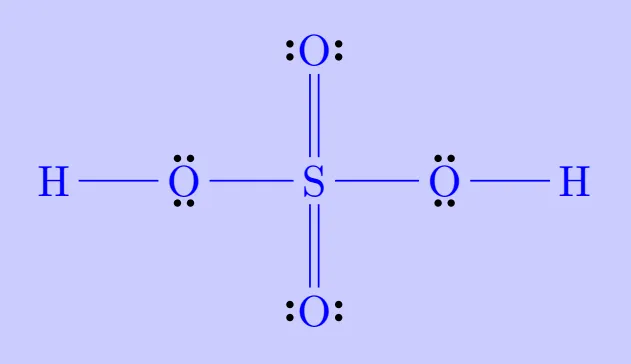
Such compounds are termed hypervalent. Their existence directly contradicts the octet rule but is well explained by the availability of additional orbitals in larger atoms.
An ionic (or electrovalent) bond is formed when one atom completely transfers one or more electrons to another atom, leading to the creation of oppositely charged ions. The bond itself arises from the electrostatic attraction between these ions. This type of bonding commonly occurs between metals, which readily lose electrons, and non-metals, which readily gain electrons.
At the core of ionic bonding lies the tendency of atoms to attain a stable outer-shell configuration. Metals generally achieve stability by losing valence electrons and forming positive ions (cations), while non-metals gain those electrons to form negative ions (anions). Once formed, these ions attract each other strongly, giving rise to an ionic compound.
Consider a typical metal–non-metal combination. The metal atom, having low ionisation energy, loses one or more electrons. The non-metal atom, with high electron affinity, accepts these electrons. As a result:
These ions arrange themselves in space so that attractive forces between unlike charges are maximised and repulsive forces between like charges are minimised. This ordered three-dimensional arrangement is called an ionic lattice.
Unlike covalent bonds, ionic bonds do not involve sharing of electrons. Instead, the bonding force is purely electrostatic and acts equally in all directions.
The formation of an ionic compound involves several energy changes:
The high lattice energy plays a crucial role in stabilising ionic compounds and compensating for the energy needed during earlier steps.
Ionic bonds exhibit several distinctive features:
Because of the nature of ionic bonding, ionic compounds generally show:
These properties clearly distinguish ionic compounds from covalent substances.
The tendency to form an ionic bond increases when:
When these conditions are satisfied, electron transfer becomes energetically favourable.
The Lattice Enthalpy of an ionic solid is defined as the energy required to completely separate one mole of a solid ionic compound into gaseous constituent ions.
This process involves both the attractive forces between ions of opposite charges and the repulsive forces between ions of like charge. The solid crystal being three dimensional; it is not possible to calculate lattice enthalpy directly from the interaction of forces of attraction and repulsion only. Factors associated with the crystal geometry have to be included.
In any molecule, the atoms are not fused together nor are they infinitely far apart. Instead, they remain separated by a definite distance at which attractive and repulsive forces are balanced. This equilibrium separation between the nuclei of two bonded atoms is called the bond length.
Bond length is therefore a fundamental structural parameter. It reflects how closely two atoms approach each other when they form a chemical bond and provides valuable insight into bond strength, molecular size, and overall geometry.
Bond length depends strongly on bond order, that is, the number of shared electron pairs between two atoms.
This trend arises because higher bond order means greater electron density between nuclei, leading to stronger attraction and reduced internuclear separation. Thus, as bond order increases, bond length decreases.
In covalent molecules, bond length can often be approximated as the sum of the covalent radii of the two bonded atoms. The covalent radius of an element is defined as half the distance between the nuclei of two identical atoms joined by a single covalent bond.
Although this method gives only approximate values, it provides a useful way to estimate bond lengths in many molecules.
In a molecule containing three or more atoms, bonds do not lie randomly in space. Instead, they adopt specific orientations that minimise repulsion and stabilise the system. The angle formed between two adjacent bonds at a central atom is called the bond angle. It is usually measured in degrees and plays a vital role in determining the overall shape and properties of a molecule.
Bond angle, together with bond length, defines molecular geometry and helps explain why molecules with the same formula can behave very differently.
Depending on the number of electron pairs surrounding the central atom, typical arrangements arise:
These ideal values are often modified in real molecules due to the presence of lone pairs or unequal bonding.
Lone pairs occupy larger regions of space than bonding pairs. As a result, they push bonding pairs closer together, reducing bond angles.
For example:
Thus, the general order of repulsion is:
lone pair–lone pair > lone pair–bond pair > bond pair–bond pair
This explains why molecules like water and ammonia have bond angles smaller than the ideal tetrahedral angle.
Several factors determine the final bond angle in a molecule:
All these influences combine to produce the observed molecular shape.
Every chemical bond represents a balance between attractive and repulsive forces. Energy is released when a bond forms, and energy is required when a bond is broken. The energy associated with breaking a bond is called bond enthalpy. It provides a quantitative measure of bond strength and plays a central role in understanding chemical reactions.
In polyatomic molecules, the same type of bond may not be identical in all positions. Therefore, instead of using individual bond energies, chemists often refer to average bond enthalpy.
Average bond enthalpy is the mean value of bond energies for a given type of bond measured over many different compounds. It represents an overall estimate rather than an exact value for any single bond.
For instance, the average C–H bond enthalpy is obtained by averaging the energies required to break all C–H bonds in a large set of molecules.
Bond enthalpy increases with bond order:
This trend arises because multiple bonds involve greater electron density between nuclei, leading to stronger attraction and making bond cleavage more difficult.
In chemical bonding, it is not enough to know that two atoms are connected; it is equally important to understand how strongly they are connected. This strength is expressed in terms of bond order. Bond order gives a numerical measure of the extent of bonding between two atoms and reflects the number of electron pairs that hold them together.
Simply stated, bond order is the number of bonds linking two atoms in a molecule or ion. It provides valuable information about bond strength, bond length, and molecular stability.
A single bond has bond order 1.
A double bond has bond order 2.
A triple bond has bond order 3.
isoelectronic molecules and ions have identical bond orders; for example, \(\mathrm{F_2}\) and
\(\mathrm{O_2^{2–}}\) have bond order 1.
\(\mathrm{N_2,\ CO\ and\ NO^{+}}\) have bond order 3.
A general correlation useful for understanding the stablities of molecules is that: with increase in bond order, bond enthalpy increases and bond length decreases.
According to the concept of resonance, whenever a single Lewis structure cannot describe a molecule
accurately, a number of structures with similar energy, positions of nuclei, bonding and non-bonding
pairs of electrons are taken as the canonical structures of the hybrid which describes the molecule
accurately.
Resonance is represented by a double headed arrow.
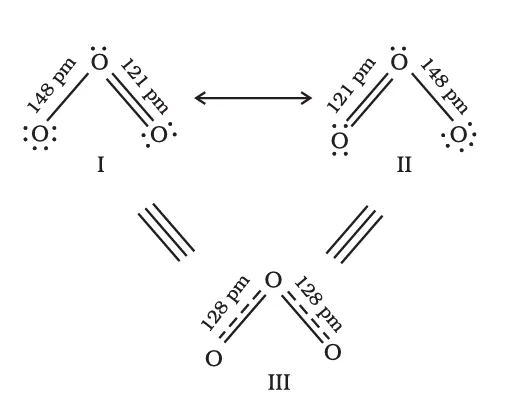
For example, in certain polyatomic ions and unsaturated molecules, experimental evidence shows that all similar bonds are of equal length and strength, even though a single Lewis structure would predict unequal bonds. This discrepancy leads to the idea of resonance.
Resonance does not imply that a molecule rapidly switches back and forth between different structures. Instead, the actual molecule exists as a resonance hybrid, which is a weighted average of all valid contributing structures.
Each resonance structure is only a hypothetical form. The real species is more stable than any individual structure and possesses properties intermediate between them.
The contributing structures are connected by a double-headed arrow (↔), indicating resonance, not equilibrium.
The resonance hybrid represents the true electronic structure. In this hybrid:
This delocalisation explains why certain molecules are unusually stable and why some bonds have lengths and strengths that do not correspond exactly to single or double bonds.
When selecting the most important resonance contributors, formal charge is used as a guide. Structures with:
are generally more significant in defining the resonance hybrid.
Resonance has several important implications:
Thus, resonance is not merely a drawing technique but a fundamental concept describing real electron behaviour.
When two atoms form a covalent bond, they share a pair of electrons. However, this sharing is not always equal. In many cases, one atom attracts the shared electrons more strongly than the other. As a result, the electron cloud becomes displaced toward one atom, giving rise to bond polarity.
The polarity of a bond refers to the unequal distribution of electron density between two bonded atoms due to differences in their electronegativities. This unequal sharing produces partial charges on the bonded atoms and leads to the formation of an electric dipole.
Electronegativity is the tendency of an atom to attract bonding electrons toward itself. When two atoms have identical electronegativities, as in a bond between identical atoms, the electrons are shared equally. Such a bond is called non-polar covalent.
If the electronegativity values differ, the more electronegative atom pulls the shared electron pair closer. Consequently:
The bond thus becomes polar covalent.
The greater the electronegativity difference, the greater the polarity of the bond. When this difference becomes very large, electron transfer may occur completely, resulting in ionic bonding.
The separation of partial charges in a polar bond creates a dipole. The strength of this dipole is measured in terms of dipole moment.
Dipole moment depends on two factors:
It is expressed as the product of charge and distance. A larger charge separation or longer bond length results in a higher dipole moment. \[\mathrm{Dipole\ moment\ (\mu) = charge\ (Q) \times\ distance\ of\ separation\ (r)}\] \[\boxed{\bbox[indigo,5pt]{\mu=Q\times r}}\]
Dipole moment is usually expressed in Debye units (D). The conversion factor is \(\mathrm{1\ D = 3.33564 \times 10–^{30}\ C\ m}\) where \(\mathrm{C}\) is coulomb and \(\mathrm{m}\) is meter.
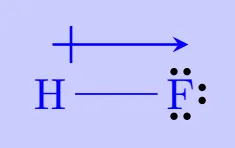
The direction of the dipole is conventionally represented by an arrow pointing from the positive end
toward
the negative end.
For example the dipole moment of HF may be represented as :
Bond polarity does not automatically mean that the entire molecule is polar. The overall molecular polarity depends on:
If bond dipoles cancel each other due to symmetrical arrangement, the molecule becomes non-polar even though individual bonds are polar. Conversely, if the dipoles reinforce each other, the molecule becomes polar.
\(\mathrm{H_2O}\) molecule has a bent structure, the two O–H bonds are oriented at an angle of 104.50.
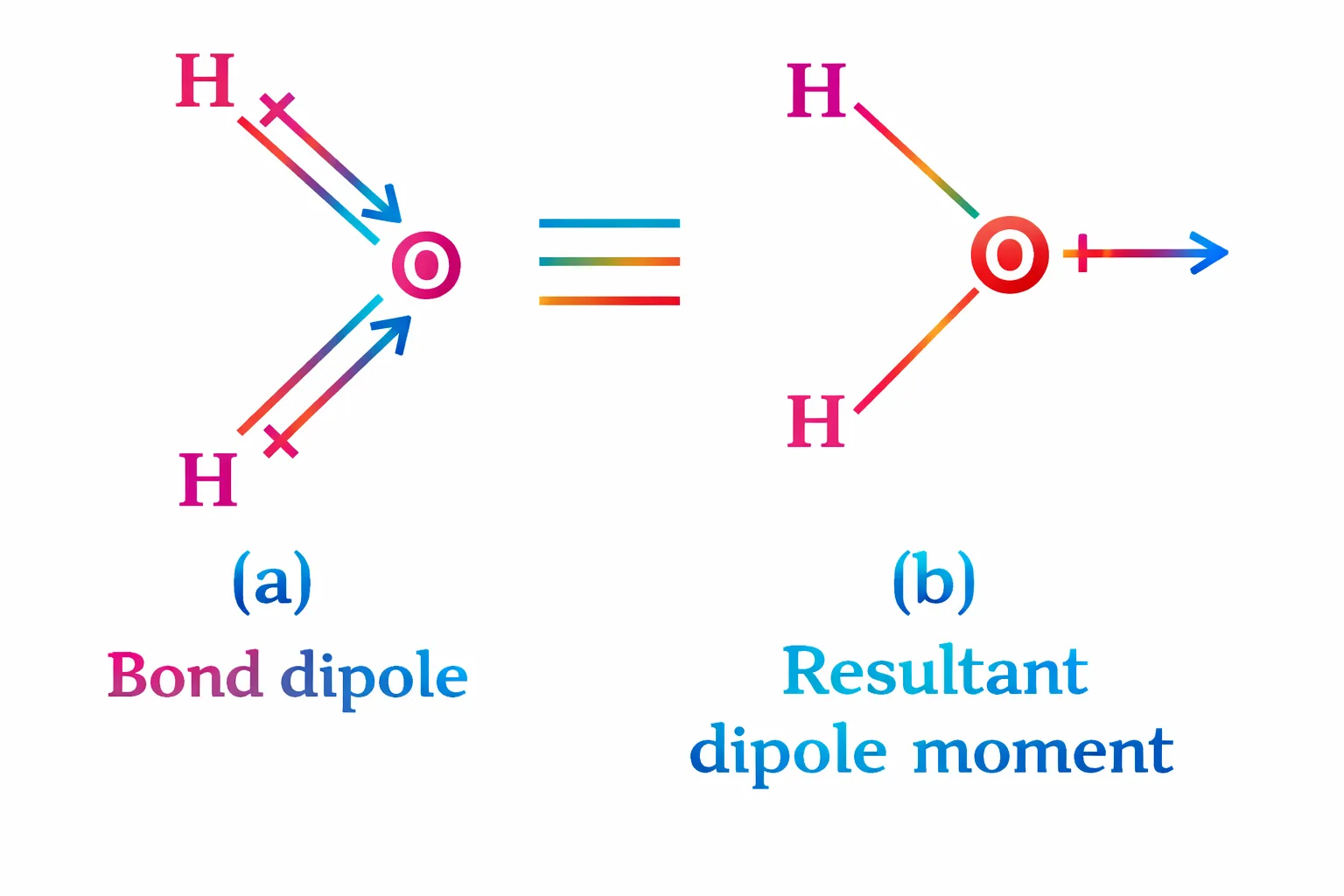
Dipole moment of each O–H bond, μ(O–H) = 1.5 D Bond angle in H₂O = 104.5°
Substituting values:
\[ \begin{aligned} μ_{net} &= 2 × 1.5 × \cos (52.25°)\\ μ_{net} &= 3 × 0.612\\ μ_{net} &≈ 1.84 D \end{aligned} \]The dipole moment of the \(\mathrm{H_2O}\) molecule is approximately 1.84 Debye.
Thus, both bond polarity and molecular shape must be considered together.
The dipole moment in case of \(\mathrm{BeF_2}\) is zero. This is because the two equal bond dipoles point in opposite directions and cancel the effect of each other.
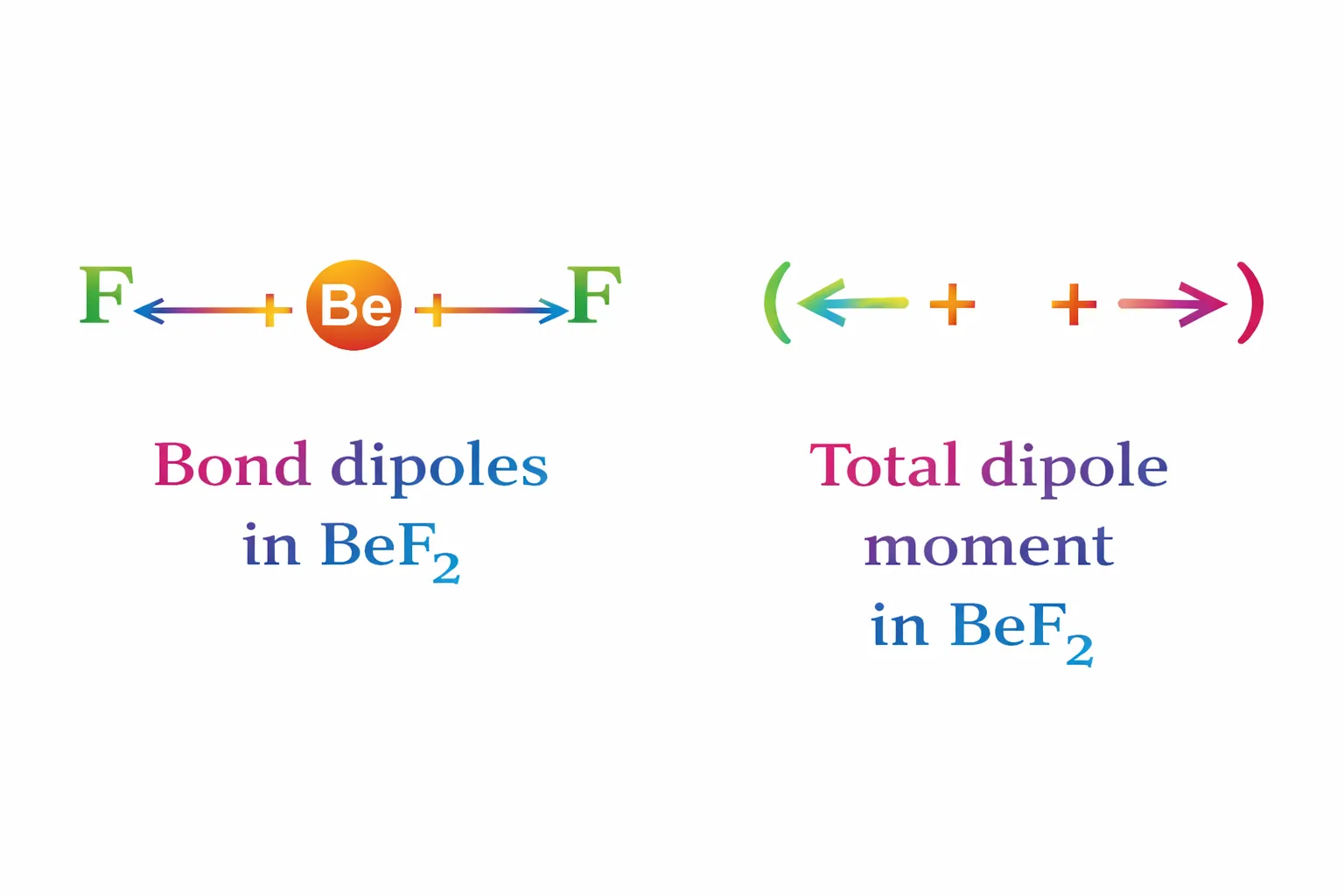
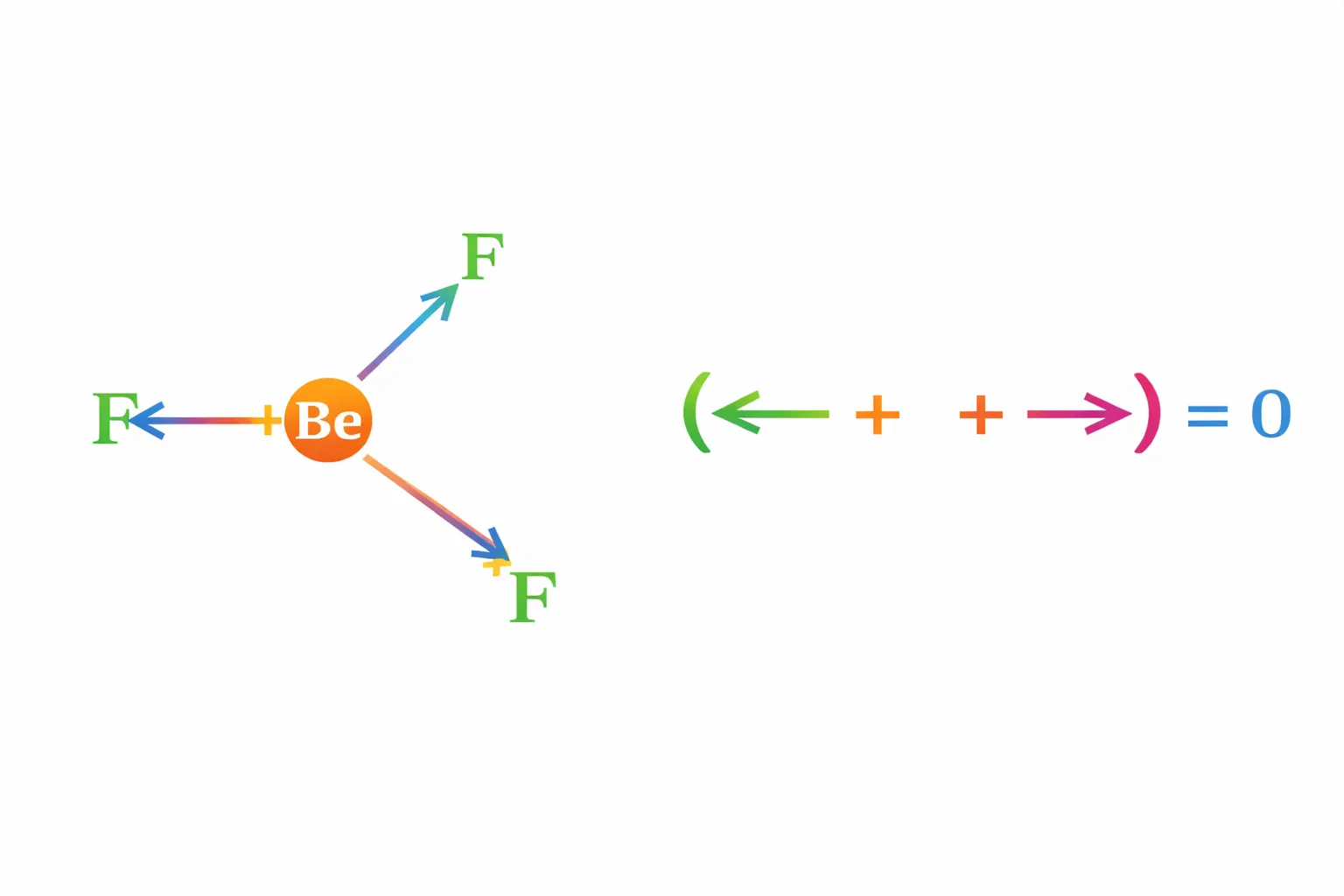
In tetra-atomic molecule, for example in \(\mathrm{BF_3}\) , the dipole moment is zero although the \(\mathrm{B – F}\) bonds are oriented at an angle of \(\mathrm{120^\circ}\) to one another, the three bond moments give a net sum of zero as the resultant of any two is equal and opposite to the third.
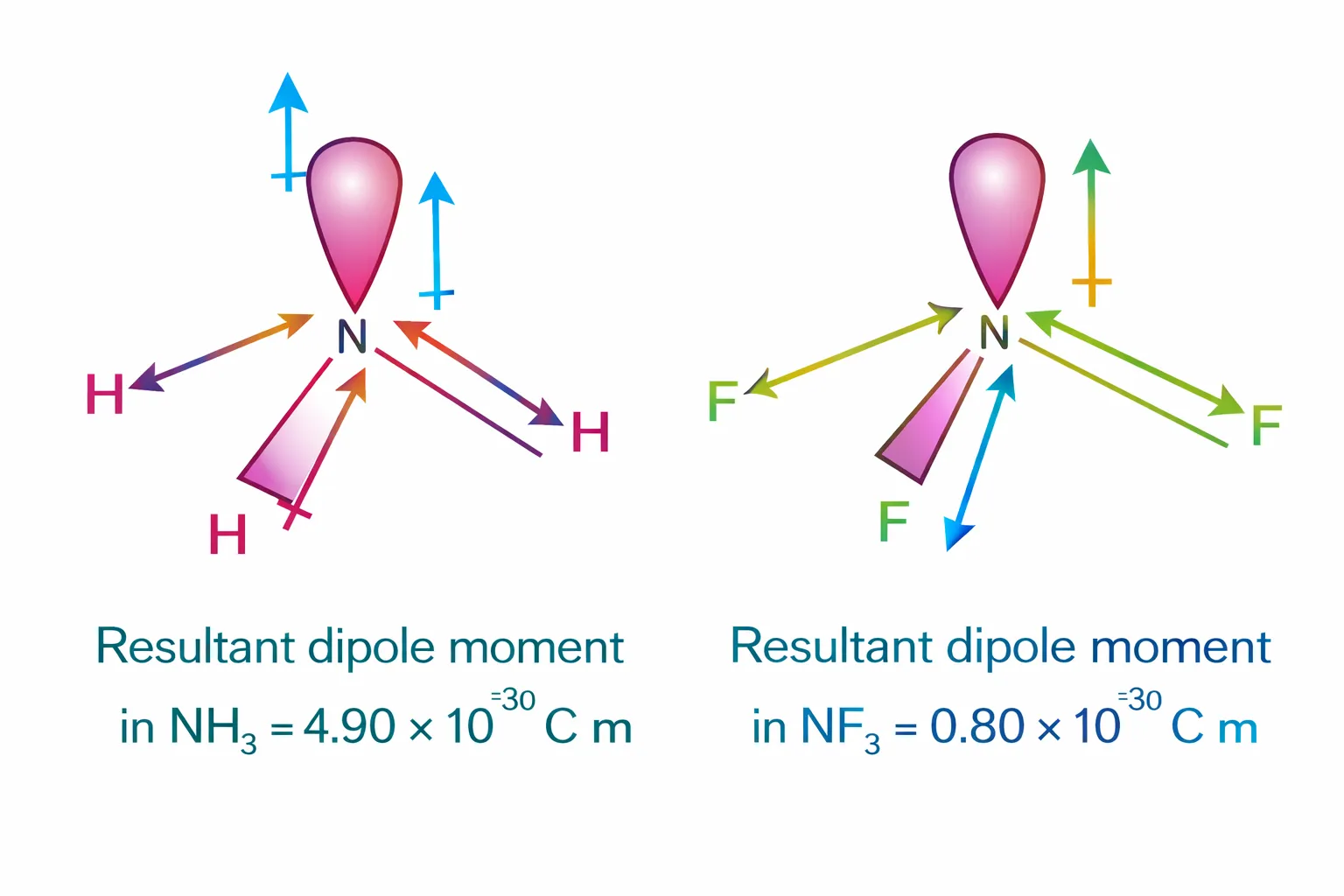
Both the molecules have pyramidal shape with a lone pair of electrons on nitrogen atom. Although fluorine
is
more electronegative than nitrogen, the resultant dipole moment of \(\mathrm{NH_3\ (4.90 × 10^{–30}\ C\
m)}\) is greater than
that of \(\mathrm{NF_3\ (0.8 × 10^{–30}\ C\ m)}\).
This is because, in case of \(\mathrm{NH_3}\) the orbital dipole due to lone pair is in the same direction
as the
resultant dipole moment of the N – H bonds, whereas in \(\mathrm{NF_3}\) the orbital dipole is in the
direction opposite
to the resultant dipole moment of the three N–F bonds.
The orbital dipole because of lone pair decreases the effect of the resultant N – F bond moments, which
results in the low dipole moment of NF3 as represented below :
Several factors influence the polarity of a bond:
Together, these factors determine whether a bond behaves as non-polar, weakly polar, or strongly polar.
Bond polarity affects many physical and chemical properties:
Polar bonds often lead to stronger intermolecular attractions compared to non-polar bonds.
Bonding is not strictly divided into purely covalent and purely ionic categories. Instead, it exists along a continuum. As electronegativity difference increases, covalent character gradually decreases and ionic character increases. Most real bonds possess some degree of both.
This perspective helps in understanding why many compounds exhibit properties intermediate between typical ionic and typical covalent substances.
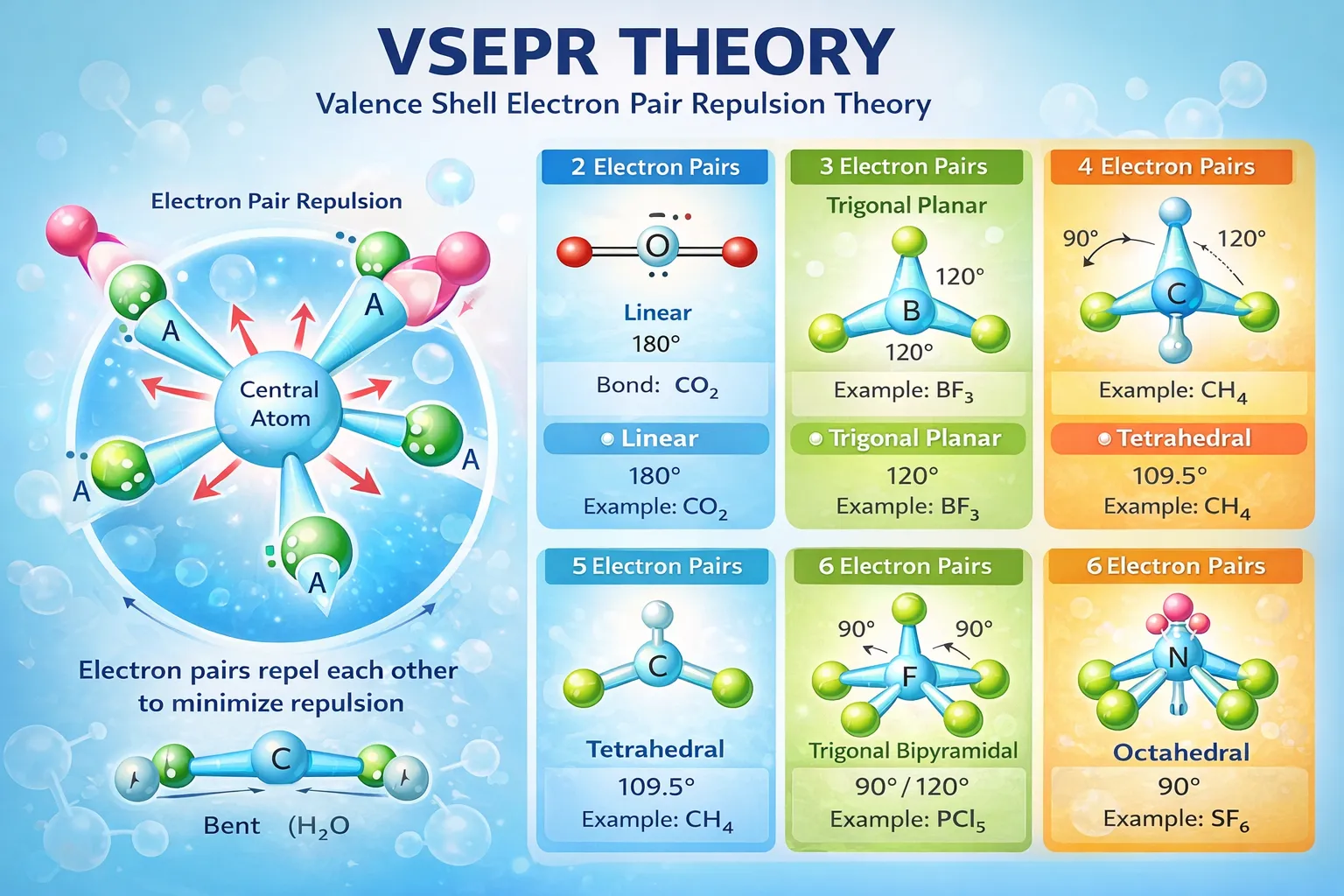
To understand why molecules adopt specific shapes, it is not enough to draw Lewis structures alone. A molecule may satisfy the octet rule and still exhibit a variety of three-dimensional arrangements. The Valence Shell Electron Pair Repulsion (VSEPR) theory provides a simple yet powerful explanation for this behaviour by focusing on the repulsion between electron pairs surrounding a central atom.
According to this theory, electron pairs in the valence shell arrange themselves in space so that mutual repulsion is minimised and separation is maximised. The observed molecular shape is therefore a direct consequence of how these electron pairs distribute themselves around the central atom.
Every pair of electrons—whether involved in bonding or present as a lone pair—occupies a region of space. Since electrons repel one another, these regions move apart as far as possible. The final arrangement corresponds to the lowest-energy configuration.
Thus, molecular geometry is governed not by atoms directly, but by electron pairs.
These electron pairs may be:
Both types influence shape, although lone pairs exert stronger repulsive effects.
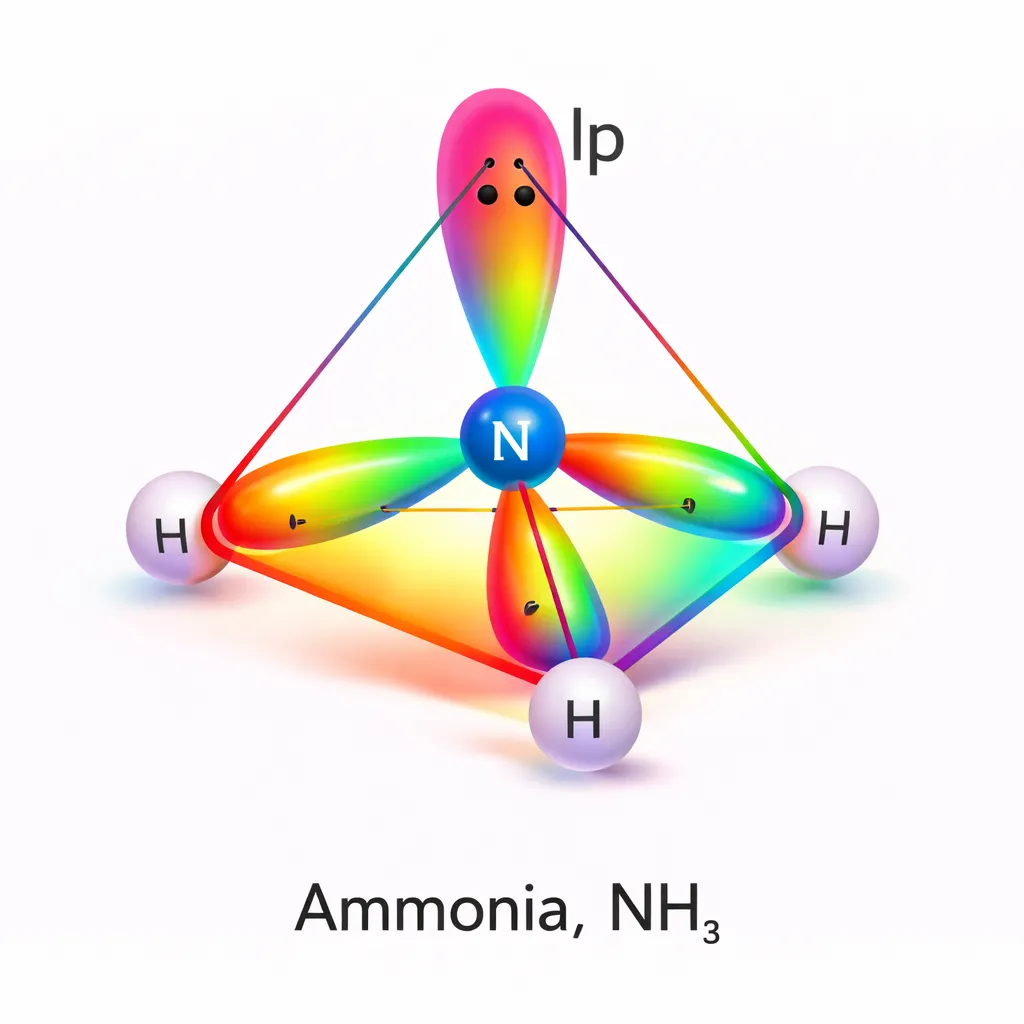
For example, four electron pairs always adopt a tetrahedral arrangement. However, if one or more of these pairs are lone pairs, the actual molecular shape changes accordingly.
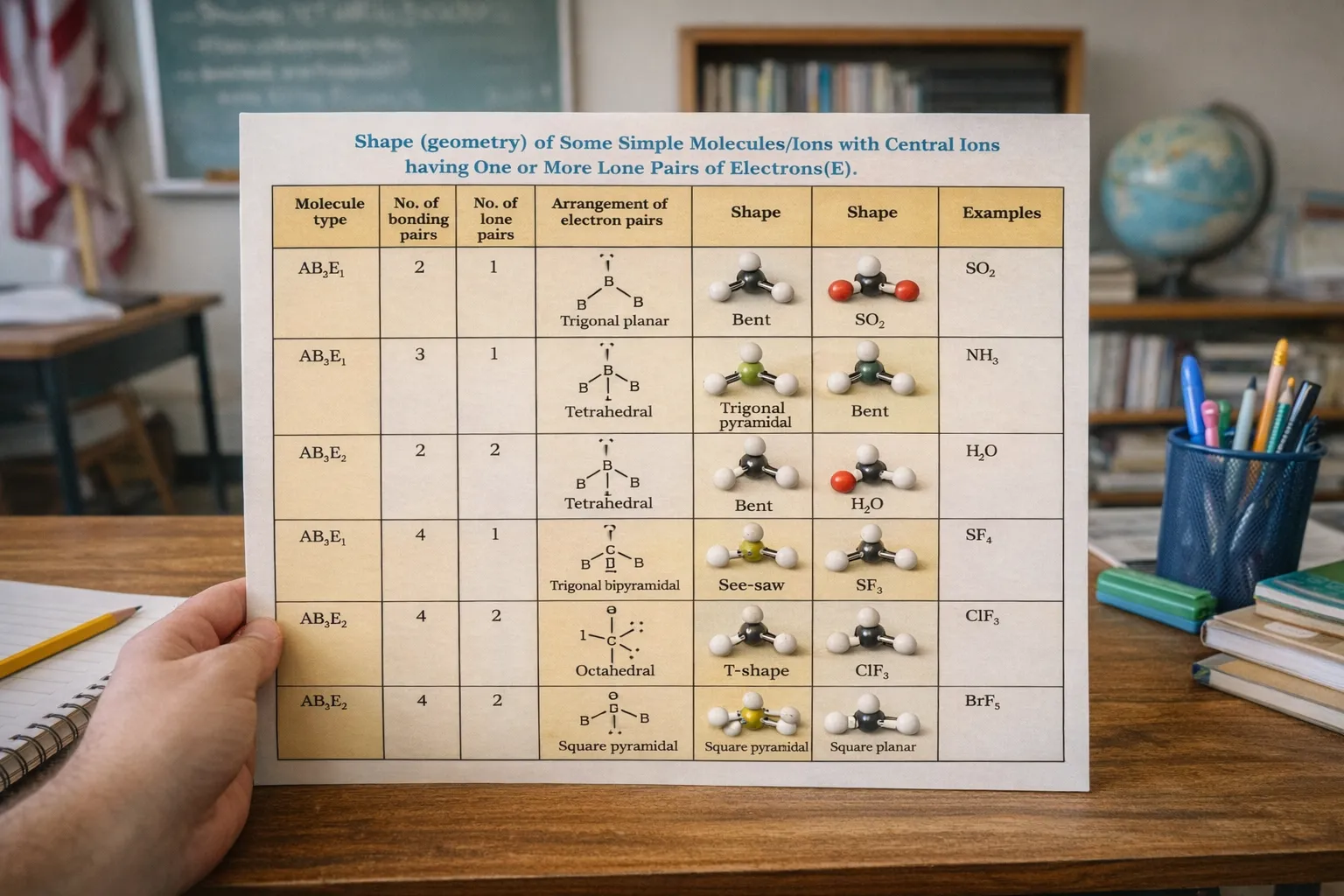
Depending on the number of electron pairs around the central atom, certain ideal geometries arise:
These represent the most widely spaced configurations possible for each case.
Lone pairs occupy more space than bonding pairs because they are attracted only by one nucleus. As a result, they repel neighbouring electron pairs more strongly. This leads to compression of bond angles.
The general order of repulsion is:
lone pair–lone pair > lone pair–bond pair > bond pair–bond pair
Because of this:
For instance, although four electron pairs favour a tetrahedral arrangement, the presence of lone pairs converts this into trigonal pyramidal or bent molecular shapes.
Double and triple bonds are treated as single regions of electron density in VSEPR theory. However, they exert slightly greater repulsion than single bonds due to higher electron concentration. This can cause small deviations in bond angles when multiple bonds are present.
Using VSEPR theory, one can predict molecular shapes by following these steps:
This approach successfully explains the geometries of many simple molecules and ions.
While VSEPR provides valuable qualitative predictions, it has certain limitations:
Despite these shortcomings, it remains an essential introductory model for understanding molecular structure.
While Lewis structures explain how atoms are connected, they do not clarify how electrons actually hold atoms together in space. The Valence Bond Theory (VBT) offers a deeper picture by describing bonding in terms of atomic orbital overlap and electron pairing.
According to this theory, a chemical bond is formed when half-filled atomic orbitals of two atoms overlap and the electrons in these orbitals pair up with opposite spins. This pairing lowers the energy of the system, leading to bond formation. The greater the overlap between orbitals, the stronger the bond produced.
Valence Bond Theory rests on a few simple ideas:
Thus, bonding is viewed as a localized interaction between specific orbitals on neighbouring atoms.
When two atoms approach each other, their atomic orbitals begin to interact. If the interaction is favourable, overlap occurs and a bond is formed.
Two main types of overlap are possible:
A sigma bond is formed along the internuclear axis and is generally stronger because of greater overlap. A pi bond results from sideways overlap of parallel orbitals and is weaker than a sigma bond.
Every single bond contains one sigma bond. Double bonds consist of one sigma and one pi bond, while triple bonds contain one sigma and two pi bonds.
One of the strengths of Valence Bond Theory is its ability to explain why covalent bonds are directional. Atomic orbitals have definite shapes and orientations in space. When these orbitals overlap, bonding occurs only in specific directions. As a result, molecules acquire characteristic geometries rather than random arrangements.
This directional nature accounts for fixed bond angles and well-defined molecular shapes.
In many molecules, simple overlap of pure atomic orbitals cannot explain observed geometries. To resolve this, Valence Bond Theory introduces hybridisation.
Hybridisation involves the mixing of atomic orbitals of similar energy on the same atom to produce a new set of equivalent hybrid orbitals. These hybrid orbitals:
Common types include linear, trigonal planar, and tetrahedral arrangements, corresponding to different hybridisation schemes. This concept successfully explains molecular shapes such as linear, planar, and tetrahedral geometries.
In the hydrogen molecule, bonding occurs by overlap of two half-filled 1s orbitals, forming a sigma bond. In methane, hybrid orbitals on carbon overlap with hydrogen orbitals to give four identical bonds arranged tetrahedrally. Such examples illustrate how Valence Bond Theory links orbital geometry with molecular structure.
Although Valence Bond Theory explains bond formation and directionality effectively, it has certain shortcomings:
These limitations led to the development of more advanced approaches, but Valence Bond Theory remains a valuable introductory model.
The formation of a covalent bond is not merely a matter of sharing electrons; it depends critically on how atomic orbitals interact in space. The orbital overlap concept explains this interaction by stating that a chemical bond is formed when atomic orbitals of two atoms overlap and allow pairing of electrons with opposite spins. This overlap lowers the energy of the system and results in a stable molecule.
In simple terms, bonding occurs because overlapping orbitals create a region of increased electron density between two nuclei, which holds the atoms together.
Each atom possesses atomic orbitals that describe the probable location of its electrons. When two atoms approach one another, their orbitals begin to interact. If half-filled orbitals from different atoms overlap effectively, electrons pair up and a covalent bond is established.
The strength of the bond depends directly on the extent of overlap:
Thus, orbital overlap provides a physical explanation for why some bonds are stronger than others.
For appreciable overlap to occur, certain conditions must be satisfied:
Only when these requirements are met does stable bond formation take place.
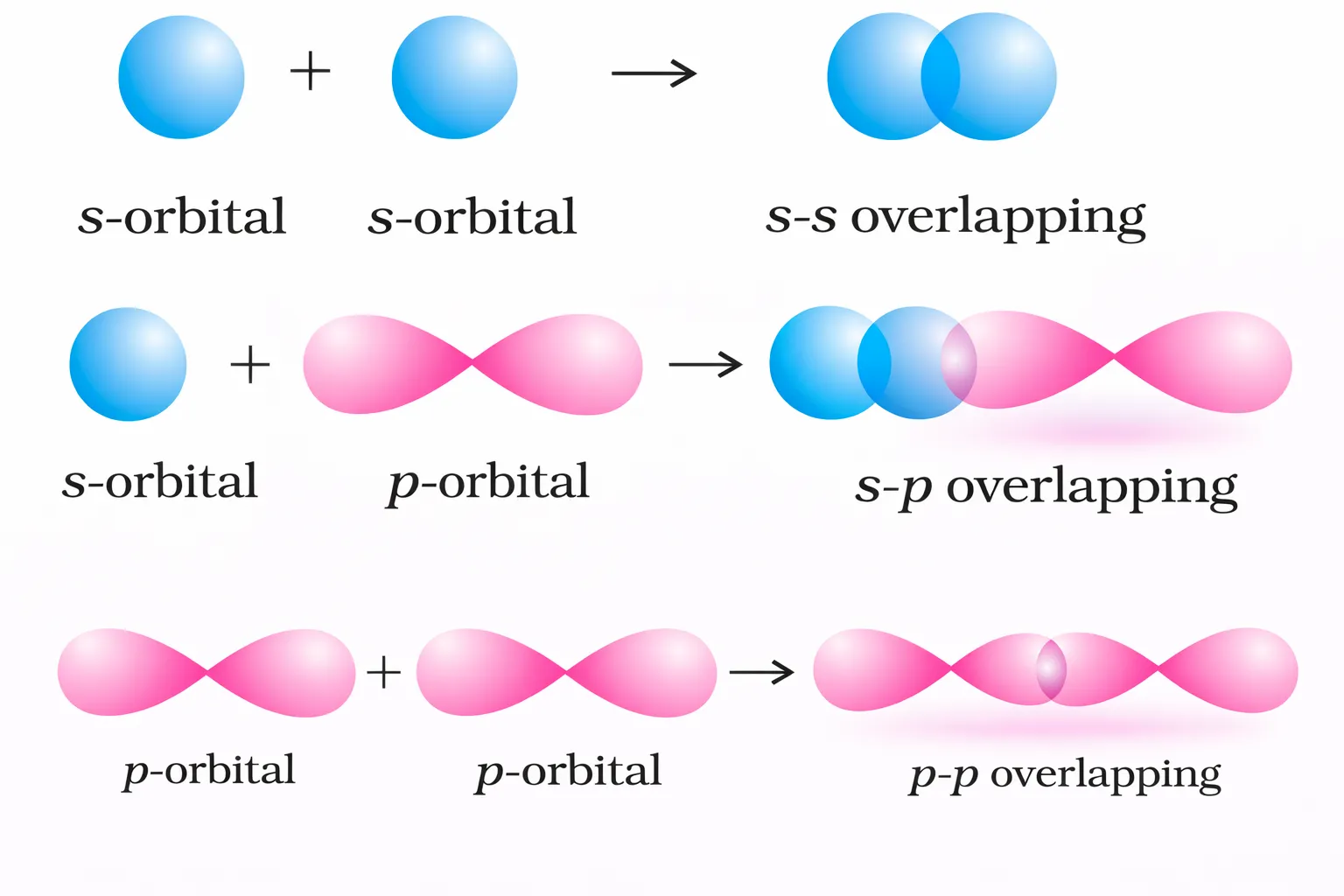
Depending on the manner in which orbitals approach each other, overlap occurs in two main ways.
Atomic orbitals have definite shapes and orientations. Because bonding requires overlap, it can occur only in specific directions. This explains why covalent bonds are directional and why molecules exhibit fixed geometries rather than random arrangements.
The concept of orbital overlap therefore forms the basis for understanding molecular shapes and bond angles.
The degree of overlap influences several important bond characteristics:
For example, multiple bonds are stronger and shorter than single bonds because they involve additional orbital overlap.
The covalent bond may be classified into two types depending upon the types of overlapping:
This type of covalentbond is formed by the end to end (headon) overlap of bonding orbitals along the internuclear axis. This is called as head on overlap or axial overlap.
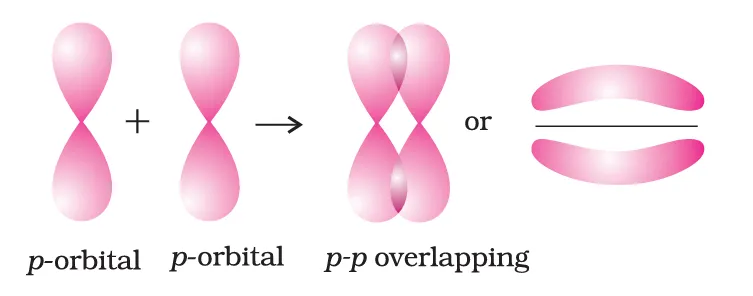
In the formation of \(\pi\) bond the atomic orbitals overlap in such a way that their axes remain parallel to each other and perpendicular to the internuclear axis. The orbitals formed due to sidewise overlapping consists of two saucer type charged clouds above and below the plane of the participating atoms.
Hybridisation describes the mixing of atomic orbitals of nearly equal energy on the same atom to form a new set of equivalent orbitals, called hybrid orbitals.
These hybrid orbitals have identical energy and shape and are oriented in specific directions in space, allowing atoms to form strong and symmetrical bonds.
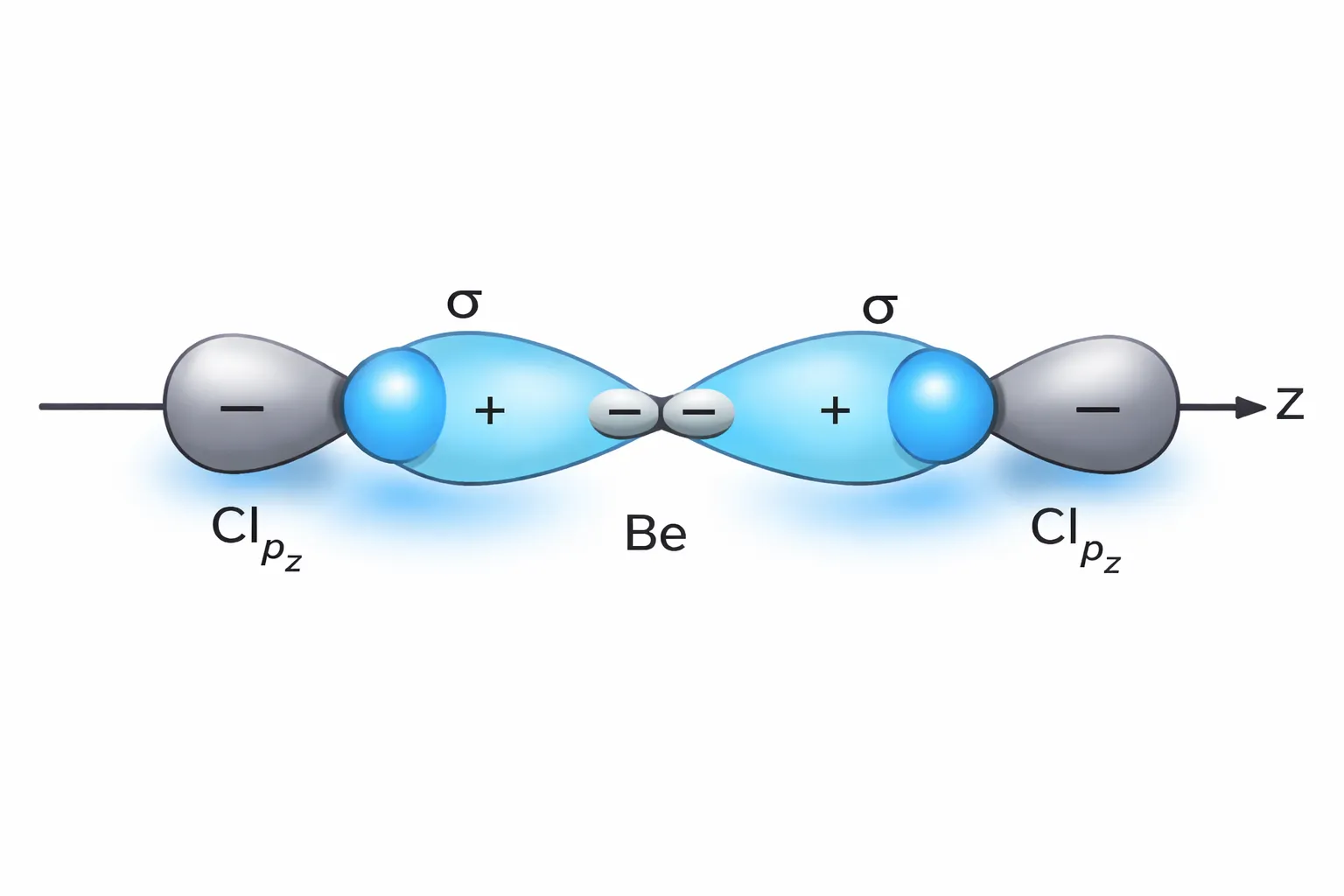
sp hybridisation occurs when one s orbital and one p orbital of the same atom mix together to form two equivalent hybrid orbitals, called sp hybrid orbitals. These hybrid orbitals are identical in energy and shape and are oriented in opposite directions in space.
This type of hybridisation is associated with a linear geometry.
The ground state electronic configuration of Be is \(1s^2\ 2s^2\). In the exited state one of the \(2s\)-electrons is promoted to vacant \(2p\) orbital to account for its bivalency. One 2s and one 2p-orbital gets hybridised to form two sp hybridised orbitals.
\[ \begin{array}{} \text{Ground State } \begin{array}{|c|c|c|} \hline { \uparrow \downarrow}\\\hline \end{array}&&\begin{array}{|c|c|c|} \hline{ \uparrow \downarrow}\\\hline \end{array}\end{array} \] \[ \begin{array}{} &\text{Exited State } \begin{array}{|r|r|r|} \hline { \uparrow \downarrow}\\\hline \end{array}&&\begin{array}{|c|c|c|} \hline{ \uparrow }\\\hline \end{array}& \begin{array}{|c|c|c|} \hline{ \uparrow }&&\\\hline \end{array} \end{array} \] \[ \begin{array}{} \text{Hybridized State } \begin{array}{|c|c|c|} \hline { \uparrow \downarrow}\\\hline \end{array}&&\begin{array}{|c|c|c|} \hline{ \uparrow }&\uparrow&&\\\hline \end{array} \end{array} \]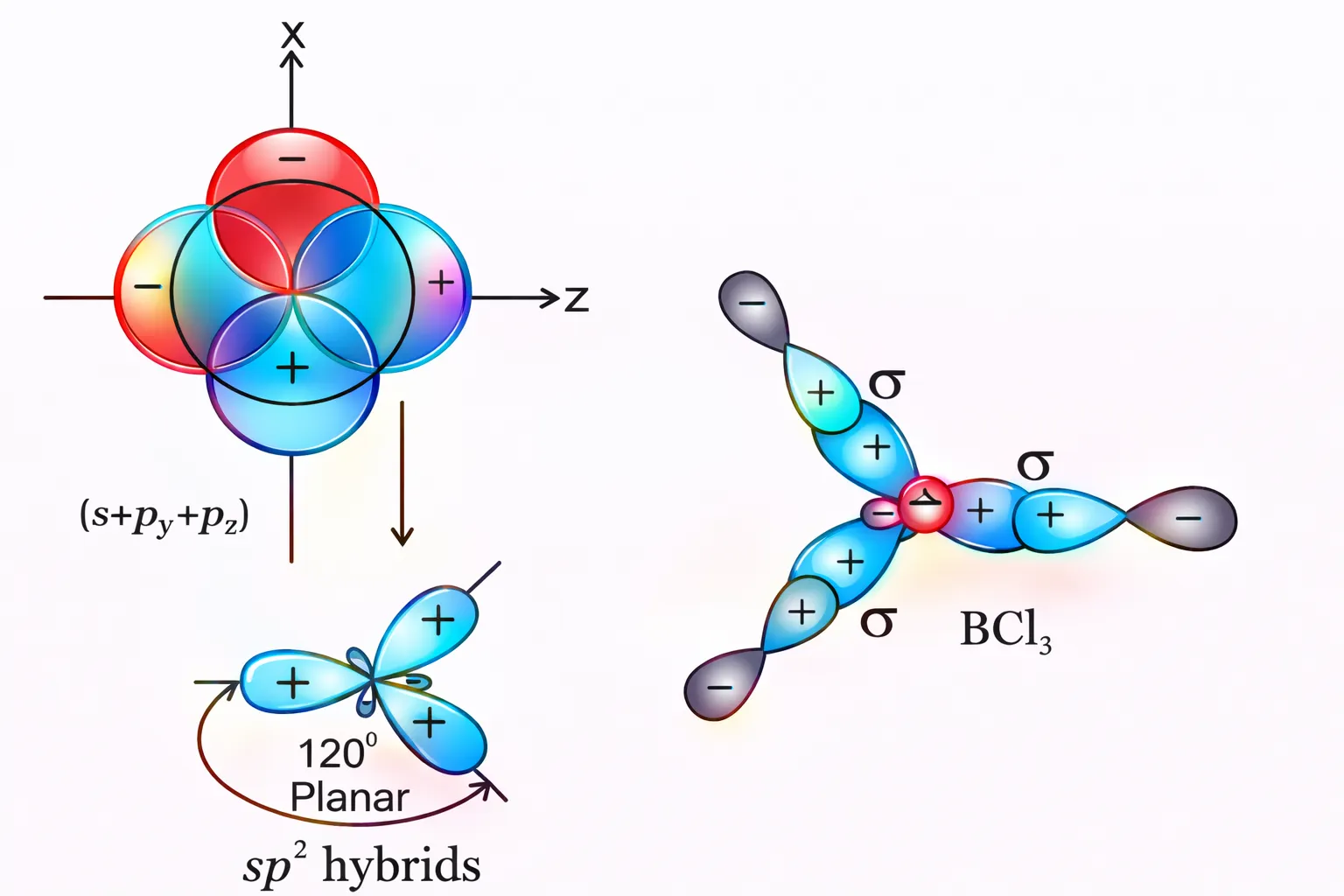
In this hybridisation there is involvement of one \(s\) and two \(p\)-orbitals in order to form three
equivalent
\(sp^2\) hybridised orbitals. For example, in \(\mathrm{BCl_3}\) molecule, the ground state electronic
configuration of
central boron atom is \(1s^22s^22p^1\). In the excited state, one of the \(2s\) electrons is promoted
to vacant \(2p\)
orbital as a result boron has three unpaired electrons.
These three orbitals (one \(2s\) and two \(2p\)) hybridise to form three \(sp^2\) hybrid orbitals.
The three hybrid orbitals so formed are oriented in a trigonal planar arrangement and overlap with \(2p\) orbitals of chlorine to form three B-Cl bonds.
\[ \begin{array}{} \text{Ground State }&&& \begin{array}{|c|c|c|} \hline { \uparrow \downarrow}\\\hline \end{array}&&\begin{array}{|c|c|c|} \hline{ \uparrow \downarrow}\\\hline \end{array}&\begin{array}{|c|c|c|} \hline{ \uparrow}&&\\\hline \end{array}\end{array} \] \[ \begin{array}{} &\text{Exited State }&&& \begin{array}{|r|r|r|} \hline { \uparrow \downarrow}\\\hline \end{array}&&\begin{array}{|c|c|c|} \hline{ \uparrow }\\\hline \end{array}& \begin{array}{|c|c|c|} \hline{ \uparrow }&\uparrow&\\\hline \end{array} \end{array} \] \[ \begin{array}{} \text{Hybridized State }& \begin{array}{|c|c|c|} \hline { \uparrow \downarrow}\\\hline \end{array}&&\begin{array}{|c|c|c|} \hline{ \uparrow }&\uparrow&\uparrow&\\\hline \end{array} \end{array} \]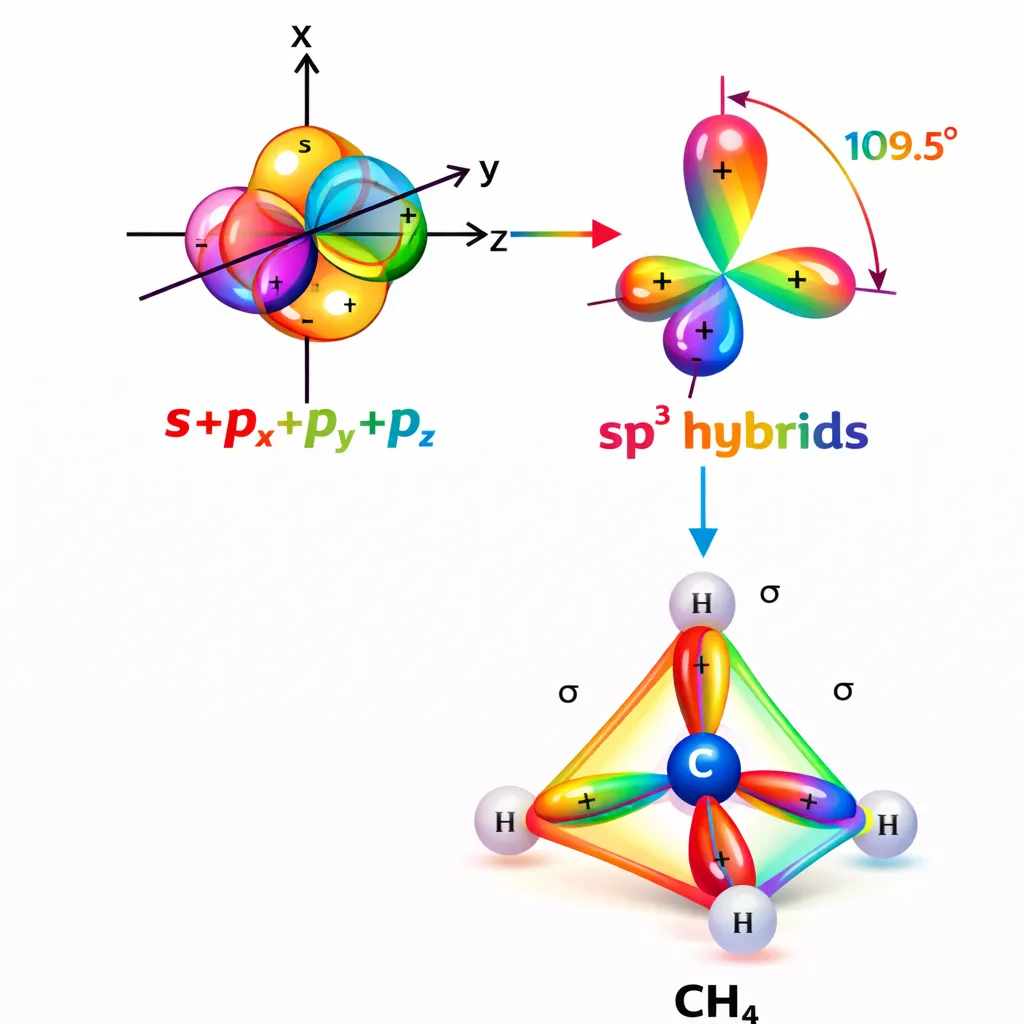
This type of hybridisation can be explained by taking the example of \(\mathrm{CH_4}\) molecule in which there is mixing of one s-orbital and three p-orbitals of the valence shell to form four \(sp^3\) hybrid orbital of equivalent energies and shape. There is 25% s-character and 75% p-character in each \(sp^3\) hybrid orbital. The four \(sp^3\) hybrid orbitals so formed are directed towards the four corners of the tetrahedron. The angle between sp3 hybrid orbital is 109.5° \[ \begin{array}{} \text{Ground State }&&& \begin{array}{|c|c|c|} \hline { \uparrow \downarrow}\\\hline \end{array}&&\begin{array}{|c|c|c|} \hline \uparrow &\downarrow\\\hline \end{array}&\begin{array}{|c|c|c|} \hline \uparrow &\uparrow&&\\\hline \end{array}\end{array} \] \[ \begin{array}{} &\text{Exited State }&&& \begin{array}{|r|r|r|} \hline { \uparrow \downarrow}\\\hline \end{array}&&\begin{array}{|c|c|c|} \hline{ \uparrow }\\\hline \end{array}& \begin{array}{|c|c|c|} \hline{ \uparrow }&\uparrow&\uparrow\\\hline \end{array} \end{array} \] \[ \begin{array}{} \text{Hybridized State }& \begin{array}{|c|c|c|} \hline { \uparrow \downarrow}\\\hline \end{array}&&\begin{array}{|c|c|c|} \hline{ \uparrow }&\uparrow&\uparrow&\uparrow\\\hline \end{array} \end{array} \]
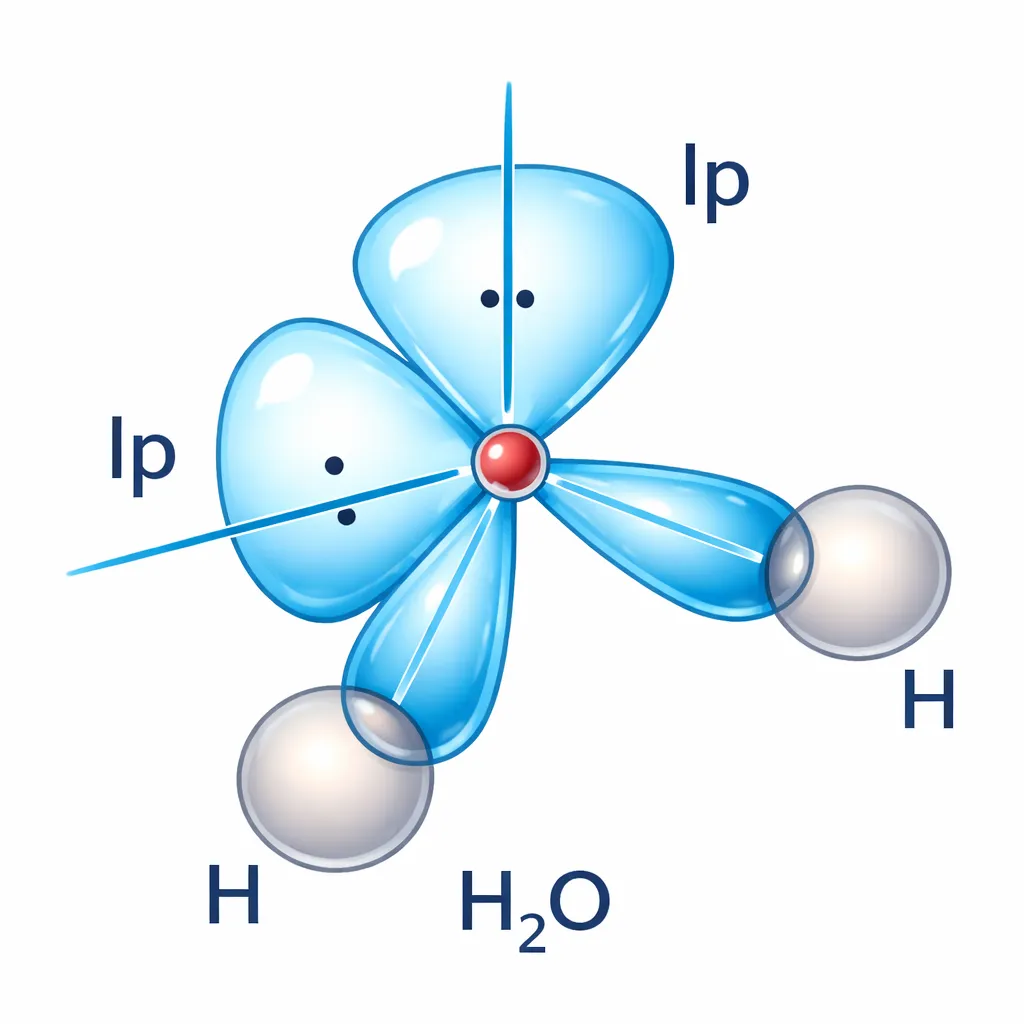
| \(P\) (Ground State) | \(\uparrow \downarrow\) | \(\uparrow\) | \(\uparrow\) | \(\uparrow\) | |||||||
| 3s | 3p | 3d | |||||||||
| \(P\) (Exited State) | \(\uparrow\) | \(\uparrow\) | \(\uparrow\) | \(\uparrow\) | \(\uparrow\) | ||||||
| 3s | 3p | 3d | |||||||||
| \(\mathrm{PCl_5}\) | \(\uparrow\color{red}\downarrow\) | \(\uparrow\color{red}\downarrow\) | \(\uparrow\color{red}\downarrow\) | \(\uparrow\color{red}\downarrow\) | \(\uparrow\color{red}\downarrow\) | ||
| \(sp^3d\) | |||||||
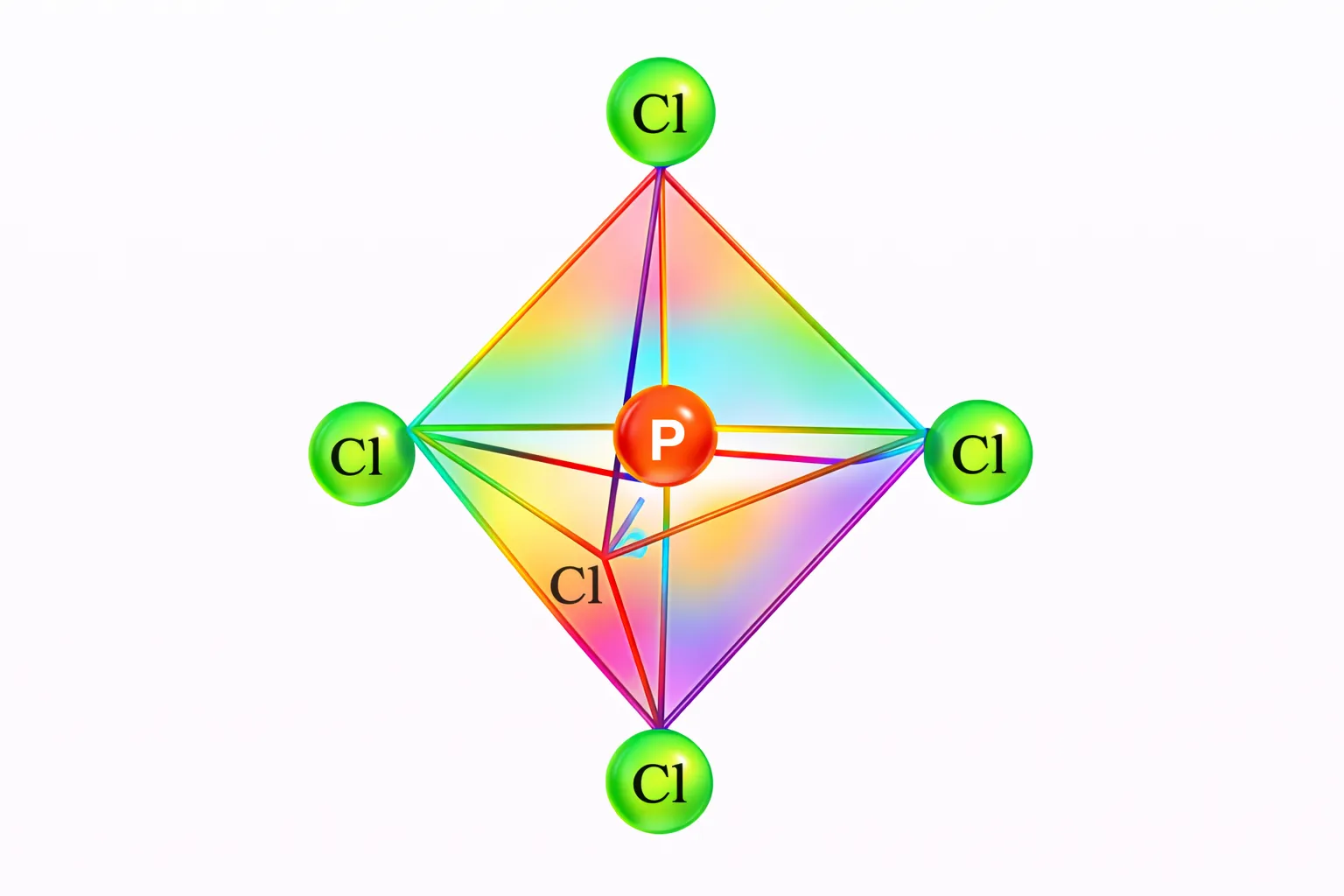
Now the five orbitals (i.e., one s, three p and one d orbitals) are available for hybridisation to yield a set of five \(sp^3d\) hybrid orbitals which are directed towards the five corners of a trigonal bipyramidal.
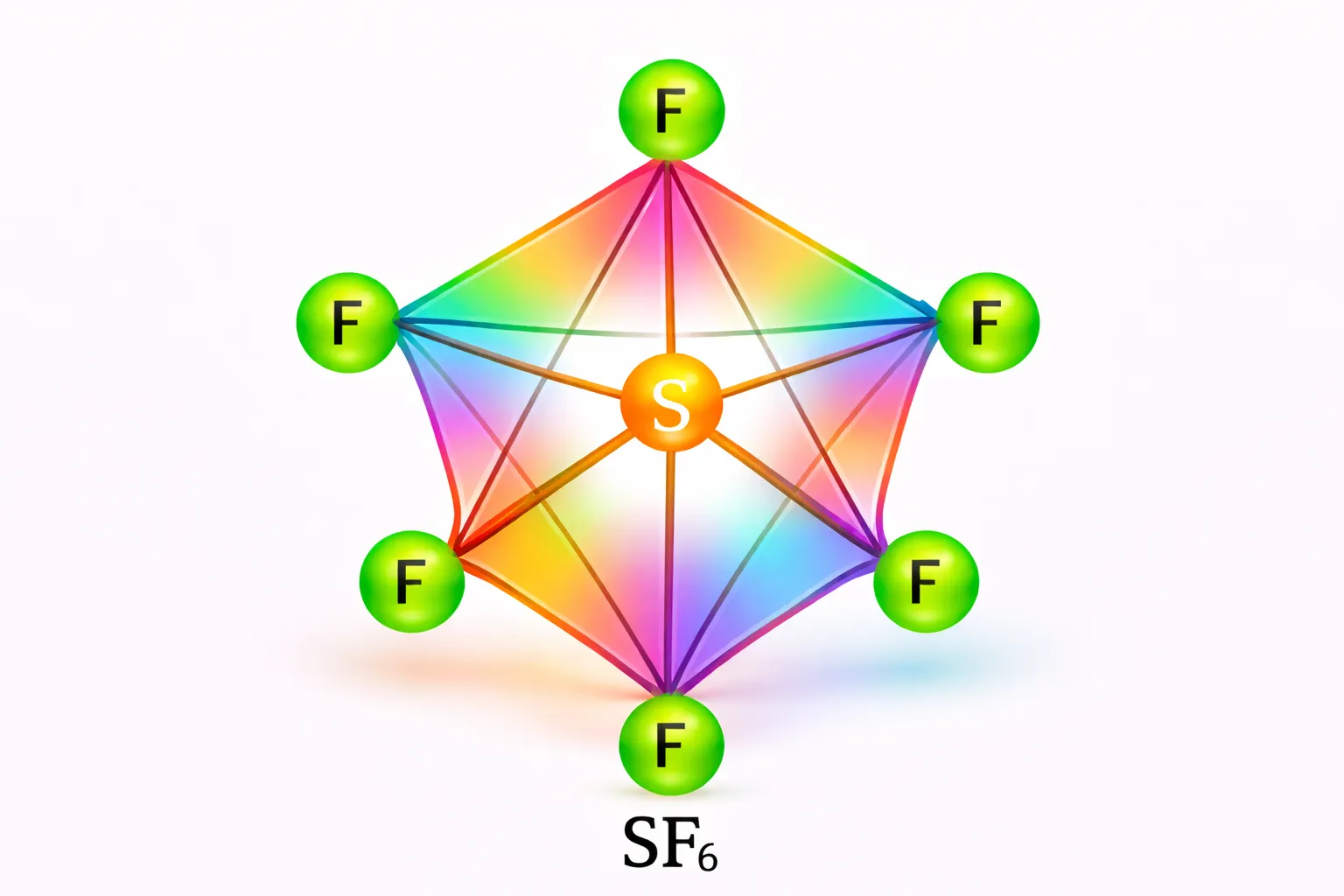
In \(SF_6\) the central sulphur atom has the ground state outer electronic configuration
\(3s2p^33p^4\). In the exited state the available six orbitals i.e., one \(s\), three \(p\) and two
\(d\) are singly occupied by electrons. These orbitals hybridise to form six new sp3d2 hybrid
orbitals, which are projected towards the six corners of a regular octahedron in \(SF_6\).
These six \(sp^3d^2\) hybrid orbitals overlap with singly occupied orbitals of fluorine atoms to
form six S–F sigma bonds. Thus \(SF_6\) molecule has a regular octahedral geometry
| \(S\) (Ground State) | \(\uparrow \downarrow\) | \(\uparrow\downarrow\) | \(\uparrow\) | \(\uparrow\) | |||||||
| 3s | 3p | 3d | |||||||||
| \(S\) (Exited State) | \(\uparrow\) | \(\uparrow\) | \(\uparrow\) | \(\uparrow\) | \(\uparrow\) | \(\uparrow\) | |||||
| 3s | 3p | 3d | |||||||||
| \(\mathrm{SF_6}\) | \(\uparrow\color{red}\downarrow\) | \(\uparrow\color{red}\downarrow\) | \(\uparrow\color{red}\downarrow\) | \(\uparrow\color{red}\downarrow\) | \(\uparrow\color{red}\downarrow\) | \(\uparrow\color{red}\downarrow\) | ||
| \(sp^3d^2\) | ||||||||
Molecular orbital (MO) theory was developed by F. Hund and R.S. Mulliken in 1932. The salient features of this theory are :
According to wave mechanics, every atomic orbital can be represented by a wave function \((\psi)\). This wave function shows the behaviour and amplitude of electron waves and is obtained by solving the Schrödinger wave equation.
For atoms with only one electron, this equation can be solved exactly. However, for molecules or atoms having more than one electron, solving the Schrödinger equation becomes very difficult. Because of this limitation, molecular orbitals cannot be obtained directly.
To deal with this problem, scientists use an approximate method called the Linear Combination of Atomic Orbitals (LCAO) .
Let us understand this using the hydrogen molecule (H₂) as an example.
In the LCAO method, molecular orbitals are formed by combining the wave functions of atomic orbitals. This combination can happen in two ways:
Mathematically, this is written as:
\[\psi_{MO}=\psi_A\pm\psi_B\]As a result, two molecular orbitals are formed:
Molecular orbitals are formed when electron waves from two atoms combine. This can happen in two ways:
When electron waves reinforce each other, a bonding molecular orbital is formed. In this case, more electron density appears between the two nuclei. This reduces repulsion between the nuclei and helps hold them together. As a result, the molecule becomes more stable, and this bonding orbital has lower energy than the original atomic orbitals.
When electron waves cancel each other, an antibonding molecular orbital is formed. Here, electron density is mostly pushed away from the region between the nuclei, creating a nodal plane (a region with zero electron density). Because of this, nuclear repulsion increases, making the molecule less stable. Antibonding orbitals therefore have higher energy than the atomic orbitals.
Although one orbital goes down in energy and the other goes up, the total energy of both molecular orbitals together remains equal to the energy of the two original atomic orbitals.
The linear combination of atomic orbitals to form molecular orbitals takes place only if the following conditions are satisfied:
Molecular orbitals of diatomic molecules are designated as \(\pi\) (sigma), \(\pi\) (pi), \(\delta\) (delta), etc.
Sigma (\(\sigma\)) molecular orbitals are formed when atomic orbitals overlap directly along the line joining the two nuclei. Because of this head-on overlap, \(\sigma\) orbitals are symmetrical around the bond axis. For example, combining two 1s orbitals or two 2pz orbitals produces a pair of \(\sigma\) orbitals: one bonding (\(\sigma\)) and one antibonding (\(\sigma^*\)).
Pi \((\pi)\) molecular orbitals arise from sideways overlap of orbitals such as \(2p_x\) or \(2p_y\).
These
orbitals are not
symmetrical around the bond axis. Instead, electron density lies above and below the internuclear
axis.
The \(\pi\) bonding orbital has concentrated electron clouds on both sides of the bond, while the
\(\pi^*\)
antibonding
orbital contains a nodal plane between the nuclei where electron density is zero.
| Antibonding | MOs | \(\sigma^*2s\) | \(\sigma^*2p_z\) | \(\sigma^*2p_x\) | \(\sigma^*2p_y\) |
| Bonding | MOs | \(\sigma2s\) | \(\sigma2p_z\) | \(\sigma2p_x\) | \(\sigma2p_y\) |
The energy levels of these molecular orbitals have been determined experimentally from spectroscopic data for homonuclear diatomic molecules of second row elements of the periodic table.
The increasing order of energies of various molecular orbitals for \(\mathrm{O_2}\) and \(\mathrm{F_2}\) is given below:
\[\boxed{\bbox[indigo,5pt]{\sigma 1s \lt \sigma 1s^* \lt \sigma 2s \lt \sigma 2s^* \lt \sigma 2p_z \lt (\pi 2p_x = \pi 2p_y ) \lt (\pi^*2p_x = \pi^* 2p_y ) \lt \sigma 2p_z^*}}\]However, this sequence of energy levels of molecular orbitals is not correct for the remaining molecules \(\mathrm{Li_2 ,\ Be_2 ,\ B_2 ,\ C_2 ,\ N_2}\). For instance, it has been observed experimentally that for molecules such as \(\mathrm{B_2 ,\ C_2,\ N_2}\), etc. the increasing order of energies of various molecular orbitals is \[\boxed{\bbox[indigo,5pt]{\sigma 1s \lt \sigma 1s^* \lt \sigma 2s \lt \sigma 2s^* \lt (\pi 2p_x = \pi 2p_y )\lt \sigma 2p_z \lt (\pi^*2p_x = \pi^* 2p_y ) \lt \sigma 2p_z^*}}\]
The important characteristic feature of this order is that the energy of \(2p_z\) molecular orbital is higher than that of \(2p_x\) and \(2p_y\) molecular orbitals.
The distribution of electrons among various molecular orbitals is called the electronic configuration of
the
molecule.
By examining the electronic configuration, we can predict the following:
If \(N_b\) is the number of electrons occupying bonding orbitals and \(N_a\) the number occupying the antibonding orbitals, then
Bond order (b.o.) is defined as one half the difference between the number of electrons present in the bonding and the antibonding orbitals i.e., \[\text{Bond order (b.o.)} = \dfrac{1}{2} (N_b–N_a )\]
Integral bond order values of 1, 2 or 3 correspond to single, double or triple bonds respectively as studied in the classical concept.
The bond order between two atoms in a molecule may be taken as an approximate measure of the bond
length.
The bond length decreases as bond order increases.
If all the molecular orbitals in a molecule are doubly occupied, the substance is diamagnetic (repelled by magnetic field). However if one or more molecular orbitals are singly occupied it is paramagnetic (attracted by magnetic field), e.g., \(\mathrm{O_2}\) molecule.
Electronic Configuration=\((\sigma 1s)^2\)
\(N_b=2\)
\(N_a=0\)
\[
\begin{aligned}
\text{Bond Order}&=\dfrac{N_b-N_a}{2}\\\\
&=\dfrac{2-0}{2}\\\\
&=1
\end{aligned}
\]
Electronic Configuration=\((\sigma 1s)^2,\ (\sigma^* 1s)^2\)
\(N_b=2\)
\(N_a=2\)
\[
\begin{aligned}
\text{Bond Order}&=\dfrac{N_b-N_a}{2}\\\\
&=\dfrac{2-2}{2}\\\\
&=0
\end{aligned}
\]
Bond Order =0, hence Molecule is nor stable and does not exist.
Electronic Configuration=\((\sigma 1s)^2,\ (\sigma^* 1s)^2,\ (\sigma 2s)^2\)
\(N_b=4\)
\(N_a=2\)
\[
\begin{aligned}
\text{Bond Order}&=\dfrac{N_b-N_a}{2}\\\\
&=\dfrac{4-2}{2}\\\\
&=2
\end{aligned}
\]
Electronic Configuration=\((\sigma 1s)^2,\ (\sigma^* 1s)^2,\ (\sigma^* 2s)^2,\ (\pi2p_x^2=\pi2p_y^2)\)
\(N_b=8\)
\(N_a=4\)
\[
\begin{aligned}
\text{Bond Order}&=\dfrac{N_b-N_a}{2}\\\\
&=\dfrac{8-4}{2}\\\\
&=2
\end{aligned}
\]
Electronic Configuration=\((\sigma 1s)^2,\ (\sigma^* 1s)^2,\ (\sigma^ 2s)^2,\ (\sigma^* 2s)^2,\ (\sigma
2p_z)^2,\ (\pi2p_x^2=\pi2p_y^2),\ (\pi^*2p_x^1=\pi^*i2p_y^1)\)
\(N_b=10\)
\(N_a=6\)
\[
\begin{aligned}
\text{Bond Order}&=\dfrac{N_b-N_a}{2}\\\\
&=\dfrac{10-6}{2}\\\\
&=2
\end{aligned}
\]
When a highly electronegative elements forms a covalent bond with Hydrogen, the electrons of covalent bond shifted towards more electronegative atoms. In this phenomenon partial positive charge is develop on Hydrogen Atom.
Positively charged Hydrogen form a bond with other more electronegative atom. This bond is known as Hydrogen bond and it is weaker than the covalent bond.
Hydrogen bond is represented by a dotted line (---). \[\mathrm{–\, –\, –\, H\delta^+\,–\,F\delta^-\, –\, –\, –\,H\delta^+\,–\,F\delta^-\,– \,– \,– H\delta^+\,–\,F\delta^-}\]
Thus
Hydrogen bond can be defined as the attractive force which binds hydrogen atom of one molecule with the electronegative atom (F, O or N) of another molecule.
There are two types of H-bonds
It is formed between two different molecules of the same or different compounds. For example,H-bond in case of HF molecule, alcohol or water molecules, etc.
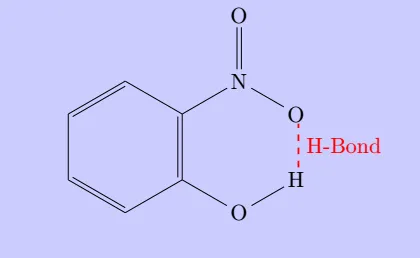
It is formed when hydrogen atom is in between the two highly electronegative (F, O, N) atoms present
within
the same molecule.
For example, in o-nitrophenol the hydrogen is in between the two oxygen atoms.
Get in Touch
Questions, feedback, or suggestions?
We'd love to hear from you.