Thermodynamics
Class 11 Notes — Complete Conceptual Guide
This chapter builds the foundation of classical thermodynamics by connecting macroscopic observations of heat and energy with the fundamental laws that govern all physical and chemical processes. It explains how energy transforms, why heat flows only in one direction, and what truly defines the concept of temperature at the deepest level.
What You Will Master
Complete conceptual guide covering all four laws of thermodynamics, thermal equilibrium, zeroth law and its significance for temperature measurement, internal energy, work and heat as path-dependent quantities, the first law of thermodynamics and its applications to various processes, specific heat capacities of gases, the second law of thermodynamics, the concept of reversible and irreversible processes, entropy as a state function, and exam-focused derivations with solved numericals.
Fundamental Relations at a Glance
where \(\Delta U\) is the change in internal energy, \(Q\) is heat supplied to the system, and \(W\) is work done by the system.
For an isobaric process: \(W = P(V_f - V_i)\). For an isochoric process: \(W = 0\).
For a monatomic ideal gas, \(C_V = \tfrac{3}{2}R\) and \(C_P = \tfrac{5}{2}R\), giving \(\gamma = 1.67\).
Why Thermodynamics Is Important for JEE & NEET
This chapter forms the backbone of physical chemistry, heat and energy theory, and the behaviour of gases under varying conditions. A strong command of this chapter directly supports Class 12 topics such as Chemical Thermodynamics, Kinetic Theory, and Heat Transfer. Most competitive exams include direct numerical questions from the first law of thermodynamics and specific heat calculations, making this one of the highest-yield chapters in the syllabus.
- First Law applications
- Work done in cyclic processes
- Adiabatic process derivations
- Mayer's relation & \(\gamma\)
- Entropy as a state function
- Zeroth Law & thermal equilibrium
- Internal energy concept
- Isothermal vs adiabatic
- Second Law & irreversibility
- P–V diagram interpretation
How Thermodynamics Appears in JEE & NEET 2026
Recent entrance exams have focused heavily on work done during different thermodynamic processes, interpretation of P–V diagrams for cyclic processes, and conceptual questions based on the second law of thermodynamics. Direct numerical problems are frequently set from the first law, Mayer's relation, and internal energy calculations for ideal gases. NEET 2026 has shown a clear rising trend of application-based questions linking thermodynamic principles to biological energy systems and chemical reactions.
Questions on entropy and irreversible processes, once rare at the Class 11 level, are increasingly appearing as single-concept MCQs, making a solid conceptual understanding of the second law essential even for NEET aspirants.
✦ Chapter Notes Begin ✦
🌌 System and Surroundings

🔹 System
System is defined as the specific portion of the universe that we choose to study or observe. It may consist of a chemical reaction mixture, a gas enclosed in a container, a solution in a beaker, or even a single substance undergoing a physical change. Everything outside this selected portion is called the surroundings, and together the system and surroundings make up the universe.
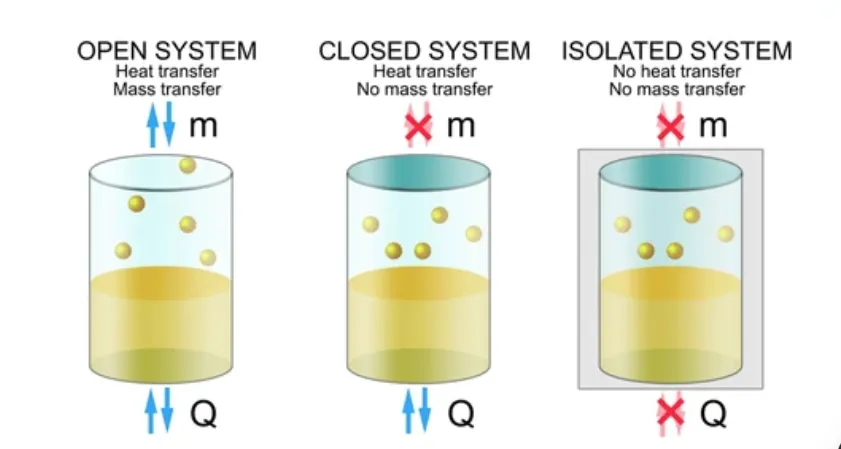
The system is separated from its surroundings by a real or imaginary boundary. This boundary determines whether energy or matter can pass between the system and the surroundings. Depending on this exchange, a system may be classified as open, closed, or isolated.
🔹 Surroundings
Surroundings refers to everything in the universe other than the system that is under study. When we select a particular portion of matter for observation and call it the system, all the remaining part, whether near or far, is considered the surroundings.
The surroundings interact with the system through the exchange of energy and, in some cases, matter. For example, when a chemical reaction takes place in a beaker, the reacting substances form the system, while the beaker, air, laboratory table, and the rest of the environment constitute the surroundings. If heat is released during the reaction, it flows from the system into the surroundings; if heat is absorbed, it flows from the surroundings into the system.
Thus, surroundings play a crucial role in thermodynamics because every thermodynamic process involves some form of interaction between the system and its surroundings.
⚙️ Types of the System
🟢 Open System
An open system is defined as a system that can exchange both energy and matter with its surroundings.
This means that not only can heat or work pass across the boundary of the system, but substances themselves can also enter or leave the system. The boundary in an open system is therefore permeable to both energy and mass.
A simple example is water boiling in an open beaker. Heat is supplied from the surroundings to the water (energy exchange), and water vapour escapes into the air (matter exchange). Since both matter and energy are being transferred, it is classified as an open system.
🟡 Closed System
A closed system is defined as a system that can exchange energy with its surroundings but cannot exchange matter.
In such a system, the boundary allows the transfer of heat or work, yet it does not permit any substance to enter or leave. The mass of the system therefore remains constant, although its energy may change due to interactions with the surroundings.
A common example is a gas enclosed in a sealed container fitted with a movable piston. Heat can be supplied to or removed from the gas, and the gas can perform work by moving the piston. However, no gas particles escape from the container.
🔴 Isolated System
An isolated system is defined as a system that can exchange neither energy nor matter with its surroundings.
In such a system, the boundary is completely non-permeable, meaning no heat, no work, and no substance can cross it. As a result, both the mass and the total energy of the system remain constant. There is absolutely no interaction with the surroundings.
A practical example that closely approximates an isolated system is a well-insulated thermos flask containing hot or cold liquid. However, in reality, perfectly isolated systems do not exist; they are only theoretical concepts.
📊 The State of the System
The state of a system refers to the condition of the system at a particular moment, as described by its measurable physical properties.
A system is said to be in a definite state when its macroscopic properties — such as pressure \((P)\), volume \((V)\), temperature \((T)\), and composition — have fixed and well-defined values. These properties are known as state variables because any change in their values leads to a change in the state of the system.
For example, if a gas is confined in a container at a certain temperature, pressure, and volume, it is considered to be in a specific thermodynamic state. If any one of these variables changes, the system moves to a new state.
The state of a system is completely specified when all its relevant thermodynamic variables are known, and it does not depend on how the system reached that condition.
🔹 State Variables
State variables are the measurable properties of a system that describe its physical condition at a given moment, including pressure \((P)\), volume \((V)\), temperature \((T)\), and amount or composition of the substance.
State variables depend only on the present condition of the system and not on the path or process by which the system reached that condition.
🔹 State Functions
State functions are those thermodynamic properties whose values depend only on the present state of the system and not on the path by which the system reached that state.
Common examples of state functions include internal energy \(\mathrm{(U)}\), enthalpy \(\mathrm{(H)}\), pressure \(\mathrm{(P)}\), volume \(\mathrm{(V)}\), temperature \(\mathrm{(T)}\), entropy \(\mathrm{(S)}\), and Gibbs free energy \(\mathrm{(G)}\).
Thus, state functions are properties that are completely defined by the state of the system itself and are independent of the process path.
⚡ The Internal Energy as a State Function
Internal energy \((U)\) is described as a state function because its value depends only on the present state of the system and not on the path followed to reach that state.
Internal energy represents the total energy contained within a system. It includes the kinetic energy of molecules due to their motion (translation, rotation, vibration) and the potential energy arising from intermolecular forces and chemical bonds.
When a system undergoes a change from an initial state to a final state, the change in internal energy is expressed as:
This change remains the same regardless of whether the process occurs in one step or through multiple intermediate steps. The first law of thermodynamics gives:
Therefore, internal energy is called a state function because its change is independent of the process path and is determined solely by the state of the system.
🛠️ Work
Work is defined as the mode of energy transfer that occurs when a force causes displacement against an opposing force. In thermodynamics, work is associated mainly with changes in volume of a system, especially in the case of gases.
When a gas expands or contracts inside a container fitted with a movable piston, work is done. If the gas expands against an external pressure, it pushes the piston outward and performs work on the surroundings. Conversely, if the surroundings compress the gas, work is done on the system.
Mathematically, when a gas expands or compresses against a constant external pressure:
where \(P_{\mathrm{ext}}\) is the external pressure and \(\Delta V\) is the change in volume. The negative sign follows the sign convention used in chemistry:
- When the system does work on the surroundings (expansion), \(w\) is negative.
- When work is done on the system (compression), \(w\) is positive.
Work is considered a path function because its value depends on how the process is carried out, not just on the initial and final states.
⚙️ Work — Detailed Mathematical Treatment
Work is defined as the energy transferred when a system undergoes displacement against an opposing force. In thermodynamics, we mainly deal with pressure–volume (P–V) work, which occurs when a gas expands or is compressed.
🔹 Expression for Work at Constant External Pressure
Consider a cylinder containing one mole of an ideal gas fitted with a frictionless piston. We know: \(\mathrm{Pressure = Force / Area}\), so \(\mathrm{Force} = P_{\mathrm{ext}} \times A\). If the piston moves by distance \(l\):
Since \(\Delta V = A \times l\), and applying the chemical sign convention:
- Compression \(\Rightarrow \Delta V < 0 \Rightarrow w > 0\) (work done on system)
- Expansion \(\Rightarrow \Delta V > 0 \Rightarrow w < 0\) (work done by system)
🔹 Work When Pressure Changes Continuously
If the external pressure changes gradually in infinitesimally small steps:
Integrating between initial volume \(V_i\) and final volume \(V_f\):
🔹 Reversible Process
In a reversible process, the external pressure differs from internal pressure by an infinitesimal amount \(P_{\mathrm{ext}} = P_{\mathrm{in}} \pm dP\). Since \(dP \cdot dV\) is extremely small:
🔹 Work in Isothermal Reversible Expansion of Ideal Gas
For an ideal gas \(P = nRT/V\). Substituting and integrating since \(nRT\) is constant at constant temperature:
Or in common logarithm form:
🔹 Free Expansion
If gas expands into vacuum, \(P_{\mathrm{ext}} = 0\), therefore \(w = 0\). In free expansion, no work is done.
🔹 Relation with First Law of Thermodynamics
Substituting \(w = -P_{\mathrm{ext}}\Delta V\) into the first law \(\Delta U = q + w\):
At constant volume \((\Delta V = 0)\):
🔥 Heat
Heat is defined as the form of energy that is transferred between a system and its surroundings due to a temperature difference.
Heat flows spontaneously from a body at higher temperature to one at lower temperature until thermal equilibrium is established. It is important to understand that heat is not a substance contained within a system; rather, it is energy in transit.
In thermodynamics, heat is represented by the symbol \(q\). According to the sign convention used in chemistry:
- \(q\) is positive when heat is absorbed by the system from the surroundings.
- \(q\) is negative when heat is released by the system to the surroundings.
Heat is classified as a path function because the amount of heat exchanged depends on the specific process by which the system changes from its initial state to its final state.
⚗️ Enthalpy (H)
In thermodynamics, most chemical reactions are performed in open containers where the pressure remains constant at atmospheric value. At constant volume, the heat absorbed equals the change in internal energy: \(\Delta U = q_V\). However, since laboratory reactions generally occur at constant pressure, it becomes necessary to define a new thermodynamic quantity — enthalpy.
Starting from the first law at constant pressure and denoting initial state as 1 and final state as 2:
Rearranging:
This suggests defining enthalpy (H):
Therefore:
Thus, at constant pressure, the heat absorbed by a system equals the change in enthalpy. For finite changes at constant pressure:
- For exothermic reactions, heat is evolved and \(\Delta H\) is negative.
- For endothermic reactions, heat is absorbed and \(\Delta H\) is positive.
At constant volume, \(\Delta V = 0\), so \(\Delta H = \Delta U = q_V\). Enthalpy is a state function defined as \(H = U + pV\), and under constant pressure conditions, the heat absorbed or released equals the enthalpy change.
📏 Extensive and Intensive Properties
🔹 Extensive Properties
An extensive property is one whose value changes with the quantity or size of the system. If the amount of substance increases or decreases, the value of an extensive property also changes. Examples: mass, volume, internal energy, enthalpy, and heat capacity.
- Depend on the amount of matter in the system.
- Additive in nature (values of parts add up to give the total).
- Change when the system is divided or combined.
- Their molar values (property per mole) become independent of quantity.
🔹 Intensive Properties
An intensive property is one whose value does not depend on the quantity or size of the system. It remains the same regardless of how much substance is present. Examples: temperature, pressure, and density.
- Independent of the amount of matter.
- Not additive in nature.
- Remain unchanged when the system is divided.
- Useful for identifying the state of a system.
🔹 Molar Property
A molar property \((\chi_m)\) is obtained by dividing an extensive property \((\chi)\) by the number of moles \((n)\):
Since it is expressed per mole, a molar property becomes independent of the amount of substance. Examples: molar volume \((V_m)\) and molar heat capacity \((C_m)\).
🌡️ Heat Capacity
Heat capacity \((C)\) tells us how much heat energy a system needs to increase its temperature by a certain amount. The two are directly related:
Here \(q\) = heat supplied, \(C\) = heat capacity, \(\Delta T\) = change in temperature. The bigger the heat capacity, the more heat is required to raise the temperature.
- the size of the system
- the amount of substance
- the nature and composition of the material
For example, water has a very high heat capacity — it takes a lot of energy to warm water, but once warm, it also cools slowly. Heat capacity is an extensive property.
🔹 Molar Heat Capacity
When heat capacity is expressed per mole, we call it molar heat capacity \((C_m)\). It is the amount of heat required to raise the temperature of one mole of a substance by 1°C (or 1 K).
🔹 Specific Heat Capacity
Specific heat capacity \((c)\) is the heat required to raise the temperature of one unit mass of a substance by 1°C (or 1 K).
🔬 Relationship between \(C_p\) and \(C_v\) for an Ideal Gas
Heat capacity at constant volume is denoted by \(C_v\) and at constant pressure by \(C_p\).
For one mole of an ideal gas, the difference between heat capacities at constant pressure and constant volume is equal to the gas constant \(R\) — this is known as Mayer's relation:
⚡ Enthalpy Change
🔹 Enthalpy Change — Definition and Derivation
In any chemical reaction, reactants are transformed into products: \(\mathrm{Reactants \rightarrow Products}\). Whenever this transformation occurs, heat may be absorbed or released at constant pressure. The heat change associated with a chemical reaction at constant pressure is called the enthalpy change of reaction, denoted by \(\Delta_rH\).
Enthalpy change of a reaction is the difference between the total enthalpy of the products and the total enthalpy of the reactants:
If \(\Delta_rH < 0 \Rightarrow\) reaction is exothermic (heat released). If \(\Delta_rH > 0 \Rightarrow\) reaction is endothermic (heat absorbed).
General Mathematical Expression
📘 Standard Enthalpy of Reaction
The enthalpy change of a reaction depends on the conditions under which it takes place. Because of this, chemists use standard conditions so that results from different experiments can be compared easily.
The standard enthalpy of reaction is the enthalpy change measured when all reactants and products are in their standard states, written as \(\Delta H^\circ\).
Standard State:
- Substance in pure form
- Pressure = 1 bar
- Usually temperature = 298 K
❄️ Enthalpy Changes during Phase Transformations
🔹 Enthalpy of Fusion
🔹 Enthalpy of Vaporisation
🔹 Enthalpy of Sublimation
Sometimes a solid changes directly into vapour without becoming liquid first. This process is called sublimation and is associated with \(\Delta_{\mathrm{sub}}H^\circ\).
⚗️ Standard Enthalpy of Formation \((\Delta_f H^\circ)\)
The standard enthalpy change for the formation of one mole of a compound from its elements in their most stable states of aggregation is called standard molar enthalpy of formation, symbol \(\Delta_f H^\circ\).
Reference state examples:
- Hydrogen → H₂(g)
- Oxygen → O₂(g)
- Carbon → C(graphite)
- Sulphur → S(rhombic)
Formation of Water
Formation of Methane
Formation of Ethanol
🔥 Thermochemical Equations
A thermochemical equation is a balanced chemical equation written together with the enthalpy change \((\Delta_rH^\circ)\) associated with the reaction. It not only tells us what substances react and form, but also how much heat is released or absorbed during the process.
Because energy changes depend on conditions and physical forms, a proper thermochemical equation always specifies the physical state (solid, liquid, gas, or aqueous) and even the allotropic form of the substances involved.
🔹 Standard Example
🔹 Important Conventions
1️⃣ Coefficients Represent Moles, Not Molecules
The stoichiometric coefficients in a thermochemical equation always refer to number of moles of reactants and products. Enthalpy change is measured per mole of reaction as written.
2️⃣ \(\Delta_rH^\circ\) Corresponds to the Written Equation
The numerical value of \(\Delta_rH^\circ\) is tied strictly to the way the equation is balanced (unit: kJ mol⁻¹). Consider:
3️⃣ Effect of Changing the Balanced Equation
Writing the same reaction with half the coefficients gives half the \(\Delta_rH^\circ\), showing enthalpy is an extensive property.
4️⃣ Reversing the Reaction Reverses the Sign
If a thermochemical equation is written in the reverse direction, the magnitude of \(\Delta_rH^\circ\) remains the same but the sign changes.
🧮 Hess's Law of Constant Heat Summation
Hess's Law is one of the most powerful ideas in thermochemistry. It arises from a simple but profound fact: enthalpy is a state function. This means the enthalpy change of a reaction depends only on the initial and final states, not on the path taken between them.
🔹 Statement of Hess's Law
If a chemical reaction occurs in several steps, the standard enthalpy change of the overall reaction equals the algebraic sum of the standard enthalpy changes of the individual steps, provided the temperature is the same.
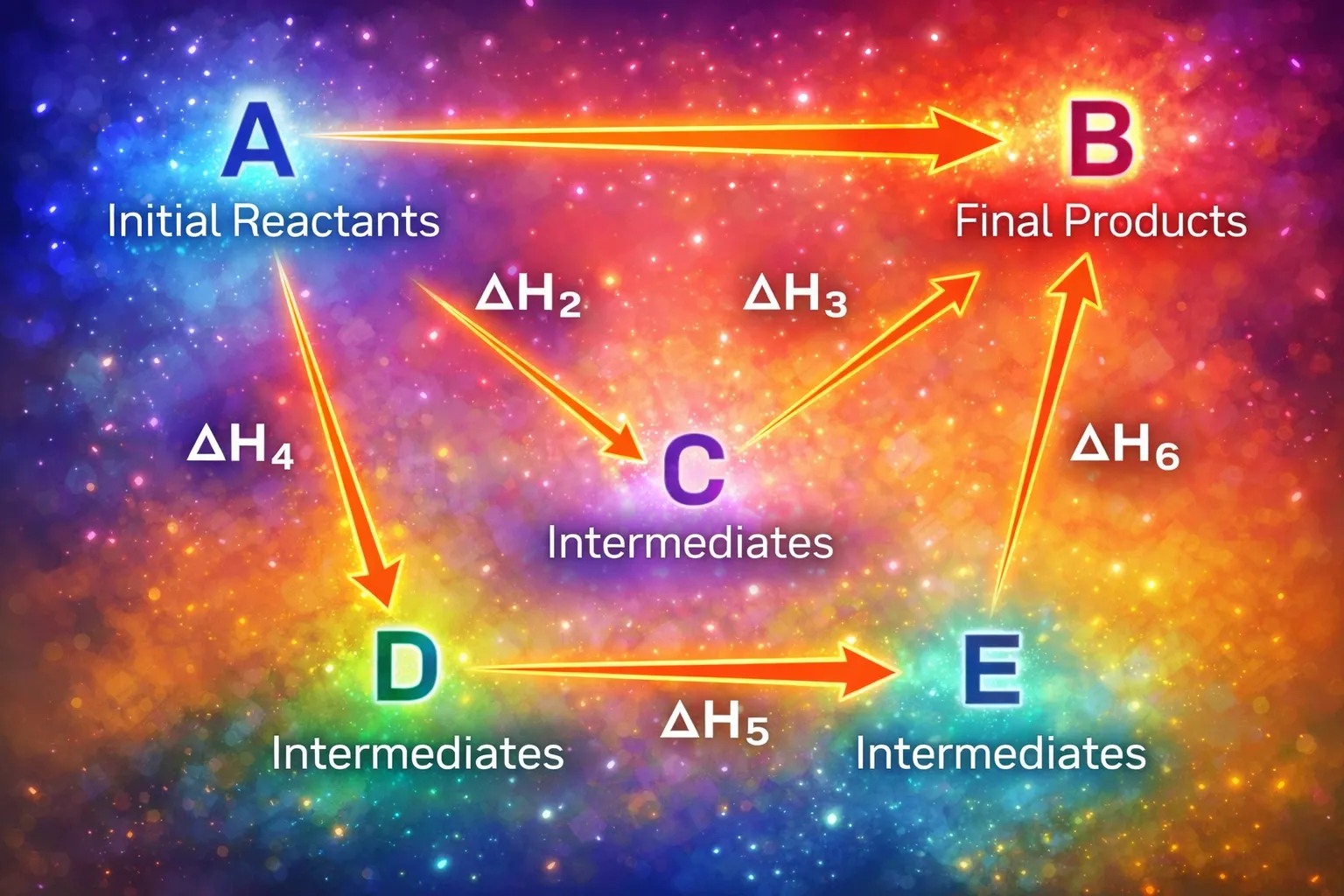
🔹 Understanding Through an Example
Consider the reaction \(C_{\text{graphite}}(s) + \frac{1}{2}O_2(g) \rightarrow CO(g)\). This is difficult to measure directly because some CO₂ always forms alongside CO.
Given Reactions
Step 1: Reverse Reaction (ii)
Step 2: Add the Equations
🔹 Key Rules to Remember
- 🔹 Enthalpy is additive
- 🔹 Reverse reaction → change sign
- 🔹 Multiply equation → multiply ΔH
- 🔹 Physical states must match
🔥 Standard Enthalpy of Combustion \((\Delta_c H^\circ)\)
The standard enthalpy of combustion is the enthalpy change when one mole of a substance undergoes complete combustion in oxygen, with all reactants and products in their standard states at 298 K and 1 bar.
- Measured per mole of the substance burned
- Combustion must be complete (no CO or soot)
- \(\Delta_cH^\circ\) is usually negative (exothermic)
- Units are typically kJ mol⁻¹
🔹 Example 1: Combustion of Butane
🔹 Example 2: Combustion of Glucose
This high energy output is the reason glucose is a primary energy source in living organisms. The human body extracts energy from glucose through cellular respiration — a controlled multistep equivalent of combustion — where energy is released gradually and stored as ATP rather than all at once as heat.
⚛️ Enthalpy of Atomization \((\Delta_a H^\circ)\)
The standard enthalpy of atomization is the enthalpy change when one mole of gaseous atoms is formed from a substance in its standard state by completely breaking all the bonds present. Since breaking bonds always requires energy, atomization enthalpy is positive in most cases.
🔹 Example 1: Dihydrogen
🔹 Example 2: Methane
All four C–H bonds are broken and all atoms are produced in the gaseous state. This large positive value shows that methane has strong covalent bonds requiring significant energy to break.
🔹 Example 3: Sodium (Metal)
- For metals: Enthalpy of atomization = Enthalpy of sublimation
🔗 Bond Enthalpy \((\Delta_{\text{bond}} H^\circ)\)
Bond enthalpy is the enthalpy change required to break one mole of a specific covalent bond in gaseous molecules to form gaseous atoms or fragments. Because bond breaking needs energy, bond enthalpy values are generally positive.
🔹 Bond Dissociation Enthalpy (Diatomic Molecules)
For diatomic molecules, bond dissociation enthalpy is straightforward because only one bond exists in each molecule.
🔹 Mean (Average) Bond Enthalpy for Polyatomic Molecules
In polyatomic molecules, even identical bonds have slightly different dissociation energies when broken stepwise. For methane \((CH_4)\), the four stepwise C–H energies are 427, 439, 452, and 347 kJ mol⁻¹. Therefore, we use an average:
🔹 Predicting Reaction Enthalpy
Example: Formation of HCl
Bonds broken = 435 + 242 = 677 kJ. Bonds formed = 2 × 431 = 862 kJ.
🧱 Lattice Enthalpy
Lattice enthalpy \((\Delta_{\text{lattice}}H^\circ)\) is defined as the enthalpy change when one mole of an ionic solid dissociates completely into its constituent gaseous ions under standard conditions.
🔹 Born–Haber Cycle for NaCl
The Born–Haber cycle breaks the formation of NaCl into hypothetical steps whose enthalpies are known experimentally:
- Step 1 — Sublimation of Na: \(Na(s) \rightarrow Na(g) \quad \Delta_{\text{sub}}H^\circ = 108.4\ \text{kJ mol}^{-1}\)
- Step 2 — Ionization of Na: \(Na(g) \rightarrow Na^+(g) + e^- \quad \Delta_i H^\circ = 496\ \text{kJ mol}^{-1}\)
- Step 3 — Dissociation of Cl₂: \(\frac{1}{2}Cl_2(g) \rightarrow Cl(g) \quad \Delta_{\text{bond}}H^\circ = 121\ \text{kJ mol}^{-1}\)
- Step 4 — Electron gain by Cl: \(Cl(g) + e^- \rightarrow Cl^-(g) \quad \Delta_{\text{eg}}H^\circ = -348.6\ \text{kJ mol}^{-1}\)
🔹 Enthalpy of Solution Relation
For NaCl: \(\Delta_{\text{sol}}H^\circ = 788 - 784 = +4\ \text{kJ mol}^{-1}\). This very small value explains why dissolving common salt produces almost no noticeable temperature change.
💧 Enthalpy of Solution \((\Delta_{\text{sol}} H^\circ)\)
The enthalpy of solution is the enthalpy change when one mole of a substance dissolves in a specified amount of solvent under standard conditions. It may be positive (endothermic) or negative (exothermic).
🔹 What Happens When an Ionic Solid Dissolves?
- Step 1 — Breaking the Crystal Lattice: \(AB(s) \rightarrow A^+(g) + B^-(g)\) — Endothermic; energy absorbed.
- Step 2 — Hydration (Solvation) of Ions: \(A^+(g) \rightarrow A^+(aq)\) and \(B^-(g) \rightarrow B^-(aq)\) — Exothermic; energy released.
🔹 Why Fluorides Are Less Soluble Than Chlorides
Fluoride ion (F⁻) is much smaller than chloride ion (Cl⁻), so ionic attraction in fluorides is stronger, making their lattice enthalpy very high. Although hydration of F⁻ is also strong, the increase in lattice enthalpy is greater, so many fluorides are less soluble: \(MgF_2\) is less soluble than \(MgCl_2\).
🔹 Example: Ammonium Nitrate
Here \(\Delta_{\text{sol}}H^\circ\) is strongly positive — highly endothermic. This is why ammonium nitrate is used in instant cold packs.
🧪 Enthalpy of Dilution
The enthalpy of dilution is the enthalpy change that accompanies the addition of extra solvent to an already prepared solution at constant temperature and pressure.
🔹 Understanding Through HCl
🔹 Calculating Enthalpy of Dilution
Subtracting the 25 aq. result from the 40 aq. result gives the heat change on further dilution:
⚡ Spontaneity
In thermodynamics, one of the most fundamental questions is not just how much energy changes, but whether a process will occur on its own. The first law of thermodynamics deals with energy conservation but does not tell us the direction in which a process will naturally proceed.
🔹 Natural Direction of Processes
- Heat always flows from higher temperature to lower temperature when left undisturbed.
- A gas released in a container spreads out to fill the entire volume.
- Carbon burns in oxygen to form carbon dioxide.
🔹 What is a Spontaneous Process?
A spontaneous process is one that has the tendency or potential to occur on its own under given conditions, without the continuous assistance of external energy.
🔹 Example: Hydrogen and Oxygen
H₂ and O₂ can be mixed and kept together at room temperature for years without noticeable change, yet the reaction is thermodynamically spontaneous. Without a spark, the activation barrier prevents the reaction from proceeding.
🔹 Important Characteristics
- Can occur without continuous external help
- Proceeds in one preferred direction
- Is generally irreversible
- Its rate may be fast or slow
- Depends on thermodynamic feasibility, not speed
🔥 Is Decrease in Enthalpy the Only Criterion for Spontaneity?
At first glance, it seems natural to believe that a process will occur spontaneously if it releases energy. Many familiar chemical reactions are exothermic and occur spontaneously:
🔹 But the Rule Fails in Some Cases
There are several reactions that are endothermic yet still spontaneous:
🔹 Classic Everyday Examples
- Melting of ice above 0°C: endothermic but spontaneous
- Dissolution of ammonium nitrate: solution becomes cold yet occurs spontaneously
- Evaporation of water at room temperature: requires heat but happens naturally
📈 Entropy and Spontaneity
Since decrease in enthalpy alone cannot always explain why a process occurs on its own, thermodynamics introduces another powerful concept — entropy (S) — which accounts for the role of disorder in determining spontaneity.
🔹 A Case Where ΔH ≈ 0 but the Process is Spontaneous
Consider a closed, isolated container divided into two compartments — left side contains gas A, right side contains gas B. When the partition is removed, the gases diffuse and form a uniform mixture spontaneously even though there may be no significant enthalpy change \((\Delta H \approx 0)\). Increased randomness is the key driving factor.
🔹 Introducing Entropy (S)
Entropy is the thermodynamic quantity that measures the degree of disorder or randomness in a system:
🔹 Entropy in Physical States
🔹 Quantitative Definition of Entropy
Experimentally, for a reversible process:
🔹 Entropy Criterion for Spontaneity
- ΔSₜₒₜₐₗ > 0 → spontaneous
- ΔSₜₒₜₐₗ = 0 → equilibrium
- ΔSₜₒₜₐₗ < 0 → non-spontaneous
⚡ Gibbs Energy and Spontaneity
Most real chemical reactions occur in open or closed systems where both enthalpy and entropy change simultaneously. In such cases, neither \(\Delta H\) alone nor \(\Delta S\) alone can reliably predict spontaneity. This leads to the concept of Gibbs energy (G).
🔹 Definition of Gibbs Energy
where \(H\) = enthalpy, \(T\) = absolute temperature (K), \(S\) = entropy. Like enthalpy and entropy, Gibbs energy is a state function and an extensive property.
🔹 Gibbs Equation
For a process at constant temperature:
🔹 Criteria for Spontaneity at Constant T and P
- ✅ If \(\Delta G < 0\) → spontaneous
- ❌ If \(\Delta G > 0\) → non-spontaneous
- ⚖️ If \(\Delta G = 0\) → equilibrium
🌡️ Role of Temperature in Spontaneity
Case 1: \(\Delta H < 0,\ \Delta S > 0\)
\(\Delta G = (-) - T(+)\) → ✅ Spontaneous at all temperatures
Case 2: \(\Delta H > 0,\ \Delta S < 0\)
\(\Delta G = (+) - T(-)\) → ❌ Never spontaneous
Case 3: \(\Delta H < 0,\ \Delta S < 0\)
Low T → spontaneous | High T → non-spontaneous
Case 4: \(\Delta H > 0,\ \Delta S > 0\)
High T → spontaneous | Low T → non-spontaneous
📜 Entropy and Second Law of Thermodynamics
The first law tells us that energy is conserved, but it does not explain why natural processes prefer one direction over another. To understand the natural direction of change, we turn to the second law of thermodynamics.
🔹 Statement of the Second Law
🔥 Example: Combustion
Heat is evolved, surrounding molecules move more randomly, total entropy increases — hence the reaction is spontaneous.
🔹 Why the Second Law is Fundamental
- Explains direction of natural processes
- Predicts feasibility of reactions
- Explains irreversibility
- Forms the basis of Gibbs free energy
- Governs engines, refrigerators, and biological systems
🧊 Absolute Entropy and Third Law of Thermodynamics
As temperature approaches absolute zero (0 K), molecular motion gradually diminishes. In the limit of absolute zero, an ideal perfectly ordered crystal would have no translational or rotational motion, minimum vibrational motion, and perfectly fixed particle arrangement — so entropy approaches its lowest possible value.
📜 Statement of the Third Law
⚠️ Limitation
The third law strictly applies only to pure crystalline solids. Solutions, glasses, supercooled liquids, and imperfect crystals may retain some residual entropy even near 0 K.
🧮 Standard Entropy and Hess-Type Relation
Once absolute entropies are known, entropy change of reactions can be calculated using:
⚖️ Gibbs Energy Change and Equilibrium
Gibbs free energy helps us answer two very practical questions: Will a reaction occur on its own? And if it occurs, how far will it proceed?
🔹 What Happens to Gibbs Energy at Equilibrium?
At equilibrium, the system has no tendency to change in either direction. Thermodynamically, this means the Gibbs energy of the system is at its minimum:
🔹 Standard Gibbs Energy and the Equilibrium Constant
Or in base-10 form:
🔹 Combined Form
This equation shows that equilibrium position depends on both enthalpy and entropy changes.
📊 What the Value of K Tells Us
🔹 When \(\Delta_r G^\circ\) is large negative
\(K \gg 1\) → products strongly favored → reaction nearly complete
🔹 When \(\Delta_r G^\circ\) is large positive
\(K \ll 1\) → reactants favored → very little product formed
🔹 When \(\Delta_r G^\circ \approx 0\)
\(K \approx 1\) → appreciable amounts of both reactants and products
🔹 Final Insight
Gibbs free energy provides the bridge between spontaneity and equilibrium. While a negative \(\Delta G\) drives reactions forward, equilibrium is reached when free energy becomes minimum and \(\Delta G = 0\). The elegant relation between \(\Delta_r G^\circ\) and the equilibrium constant allows chemists to predict how far reactions will proceed and how temperature influences product yield. Thus, Gibbs energy stands at the heart of chemical thermodynamics, governing both the direction and the extent of chemical change.
✦ End of Chapter 5 ✦